Running predictions



Running predictions |
|
|
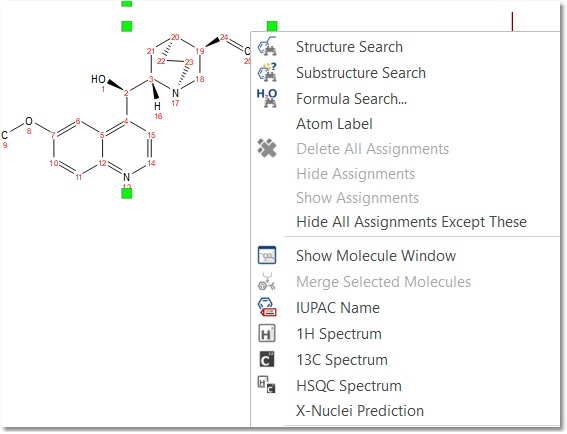
Carrying out of predictions in Mnova with NMRPredict Desktop is very easy. Just load a molecule into Mnova and right click on it to select the desired prediction (or follow the 'Prediction' menu):
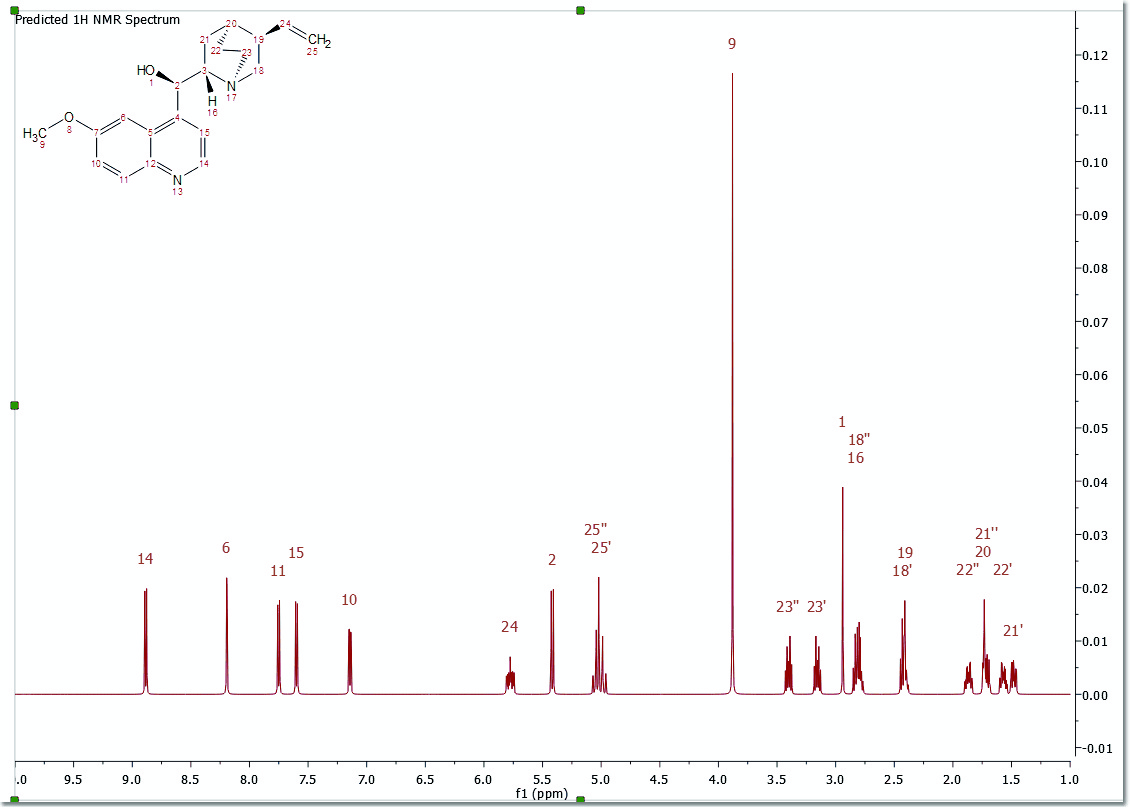
After that, you will obtain the desired predicted spectrum using a rigorous full quantum mechanics method taking into account any second order effect present in the predicted spin(s) systems(s):
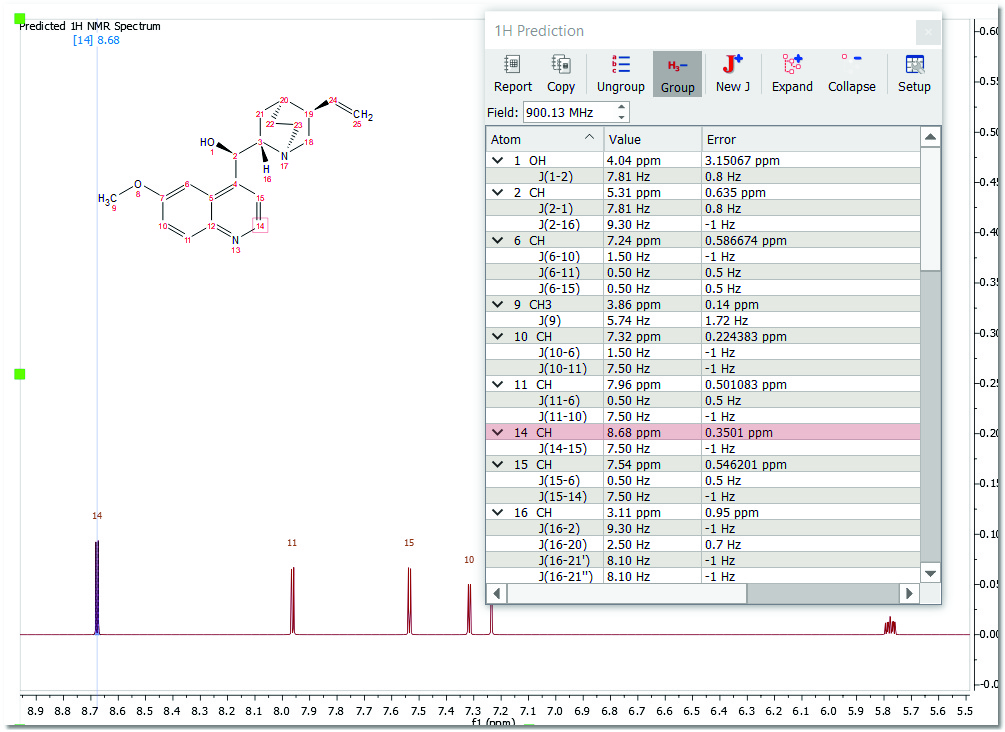
This predicted spectrum can be analyzed as a real one (e.g. it can be integrated, etc). The user can also visualize and edit the chemical shift and coupling constants values from the tables of the predicted spectrum by following the menu 'View/Table/1H Prediction'. Please bear in mind that the peak labels and the multiplet boxes will show the applicable atom numbers of the prediction.
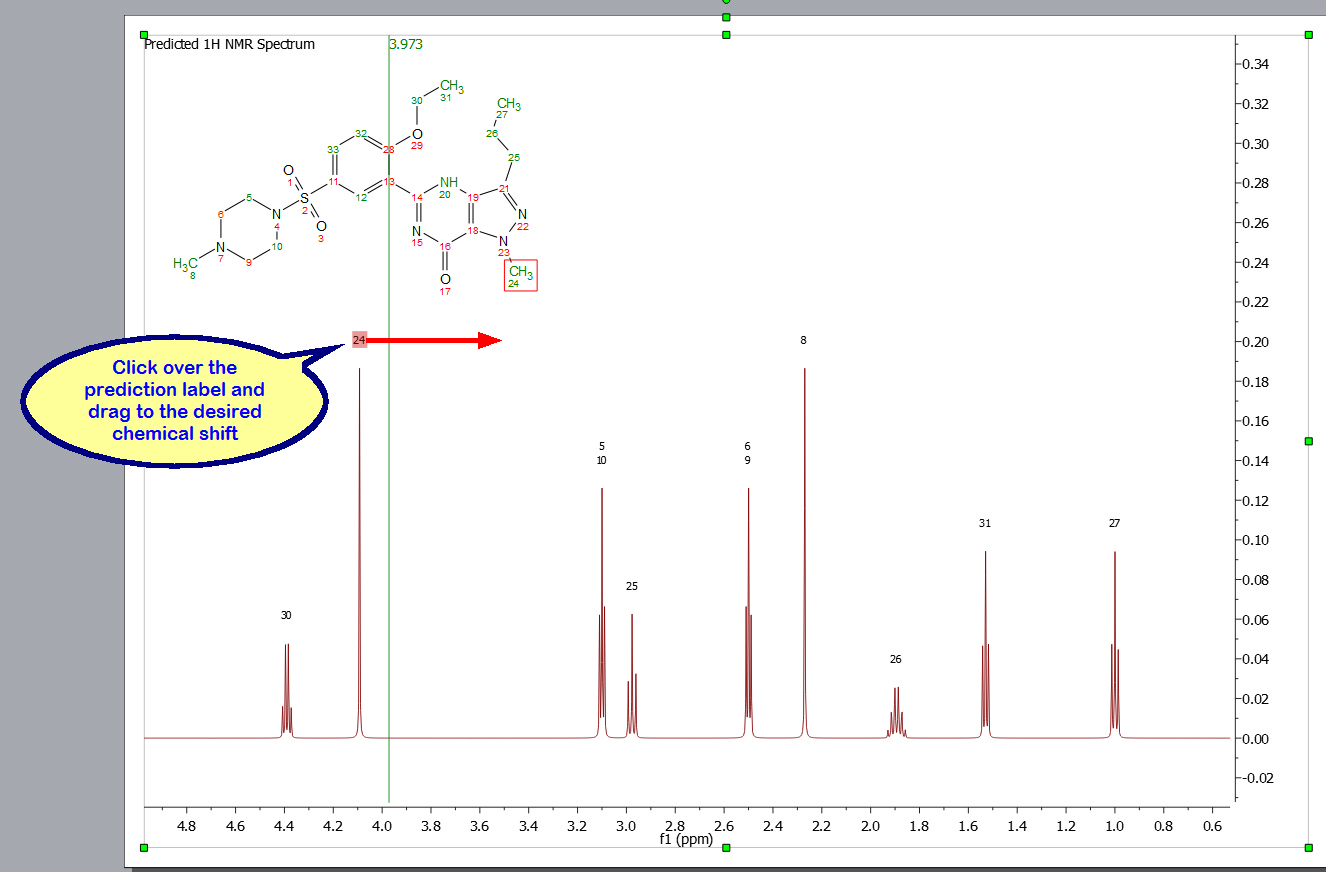
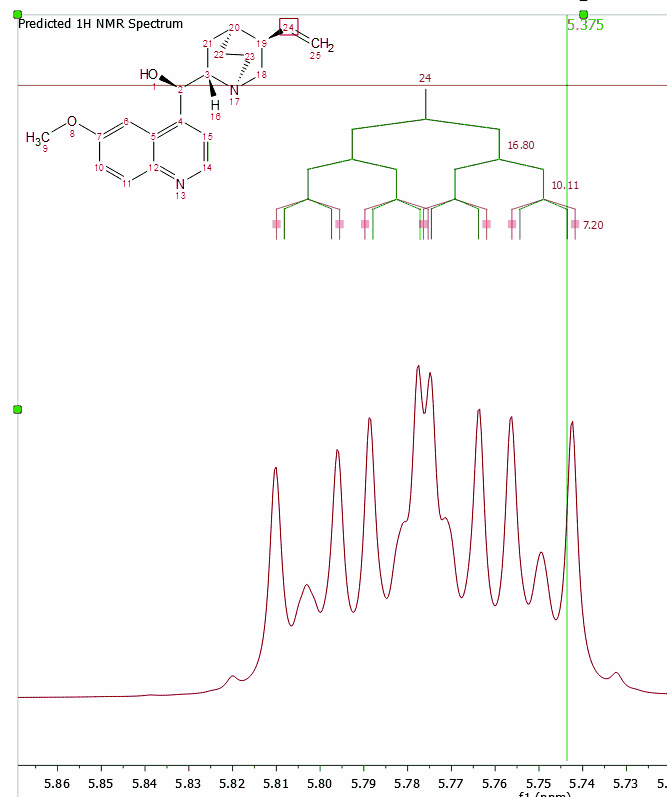
Click&drag over the prediction label to graphically modify the chemical shift of the predicted signals:
You can change the predicted coupling constants by showing the multipet tree from the 'Prediction Properties' dialog box and clicking&dragging:
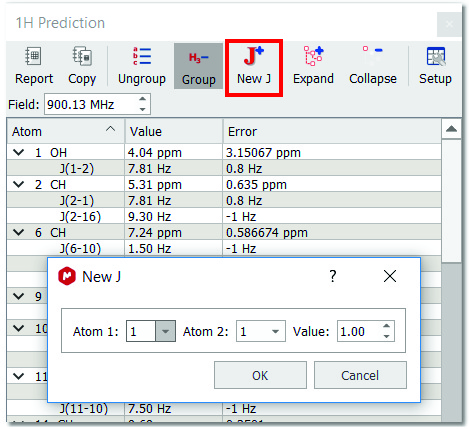
Click on the on the 'New J' button
After that, the 'New J' dialog box will be displayed and you will only need to select the atoms involved in the coupling and the value of the new coupling constant. Click on OK and the new coupling constant will be added to the spectrum and also to the prediction table.
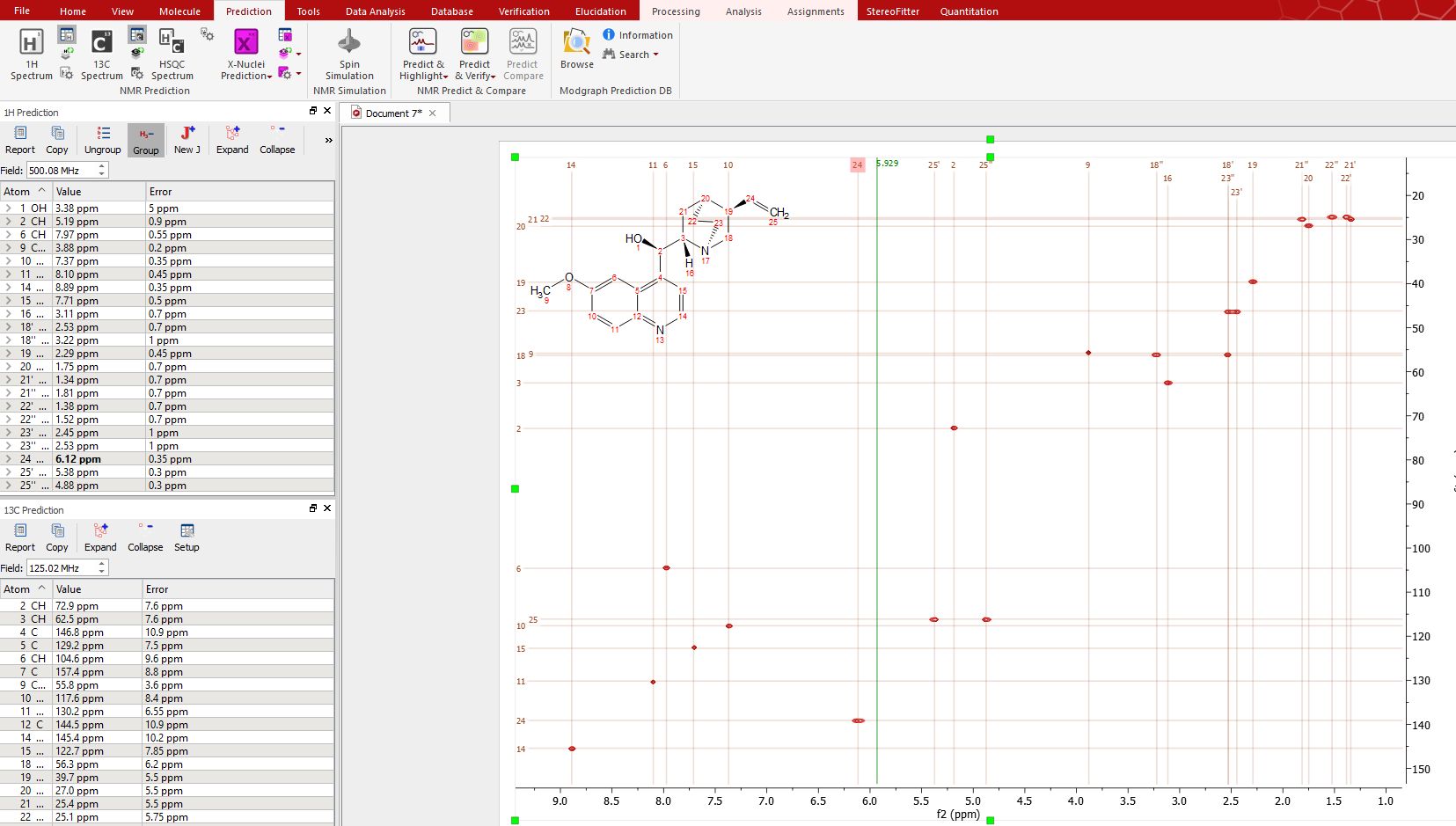
Finally, to know the "predicted assignment", just hover the mouse over the desired atom, (so that it becomes highlighted in a red box) and the corresponding signal in the spectrum will automatically be highlighted in blue; or vice versa (hovering the mouse over the peak will highlight the applicable atom on the molecular structure). You will also get highlights in the applicable row of the 'Prediction table':
Please bear in mind that NMR Predict Desktop also includes the calculation of heteronuclear (HF, HP, CF and CP) couplings:
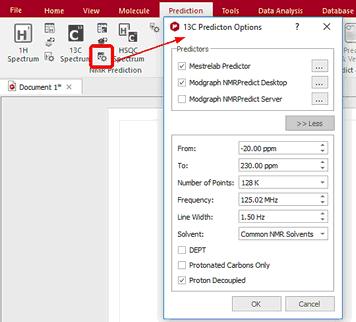
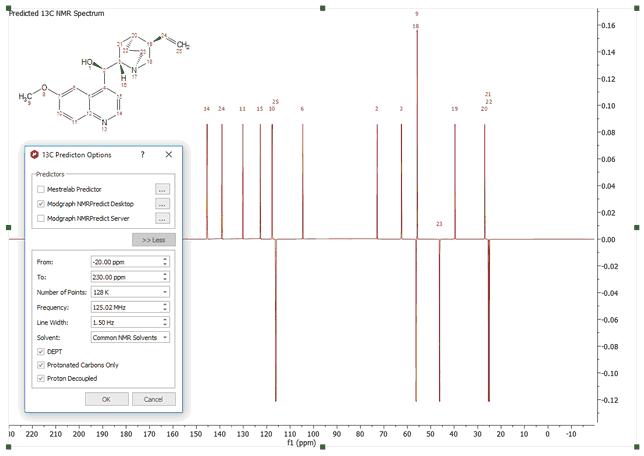
If you need the 13C-NMR predicted spectrum, follow the same instructions but selecting the options for '13C' (be sure that you select the correct icon for 13C, red square in the figure below). As you can see in the '13C Predictor Option', you can select the limits, the number of points, the frequency and the line width of the spectrum. You can predict 'DEPT' (CH and CH3 up and CH2 and Quaternary carbons down) and spectra with only protonated carbons (DEPT 45) by checking the applicable boxes. If you want to get a DEPT 135, you must check both boxes.
Here you can see an example of a DEPT 135 prediction:
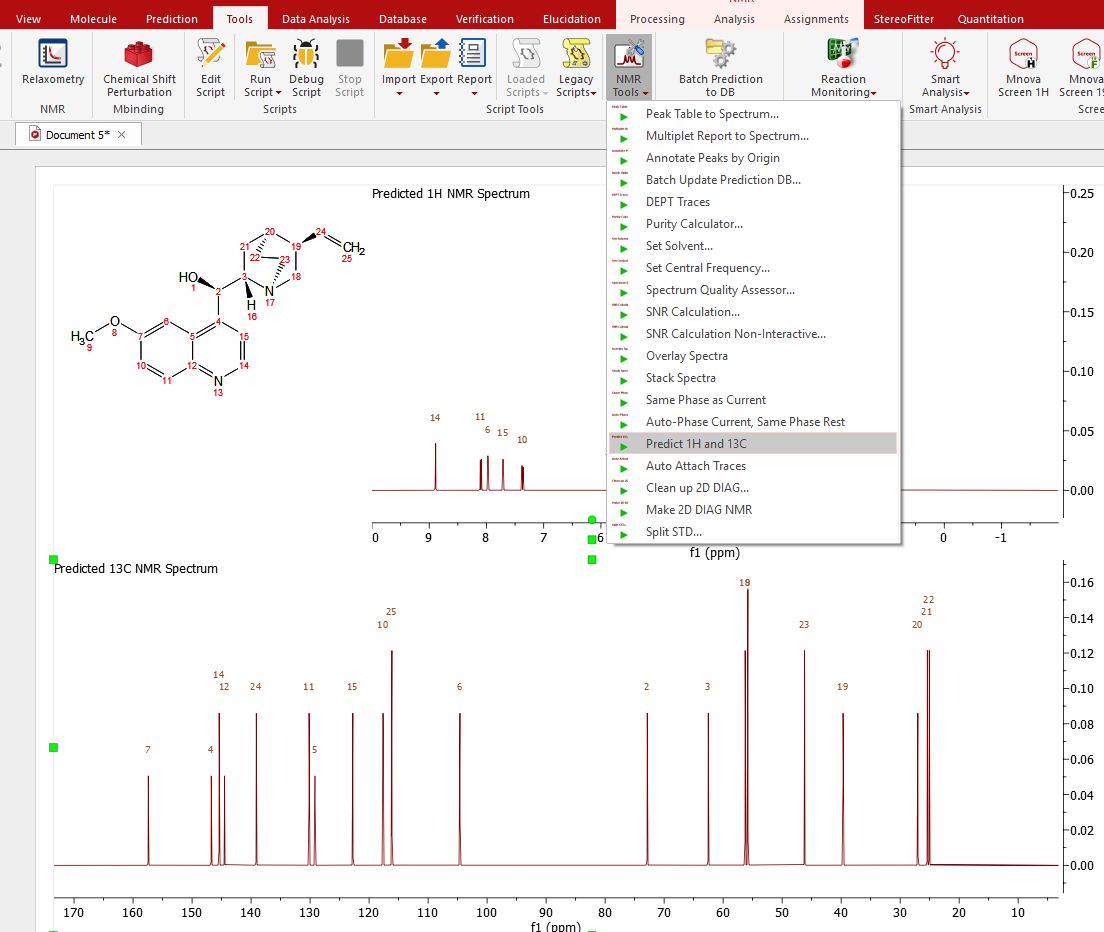
Follow the menu 'Tools/NMR Tools/Predict 1H and 13C' to get both predictions in the same page:
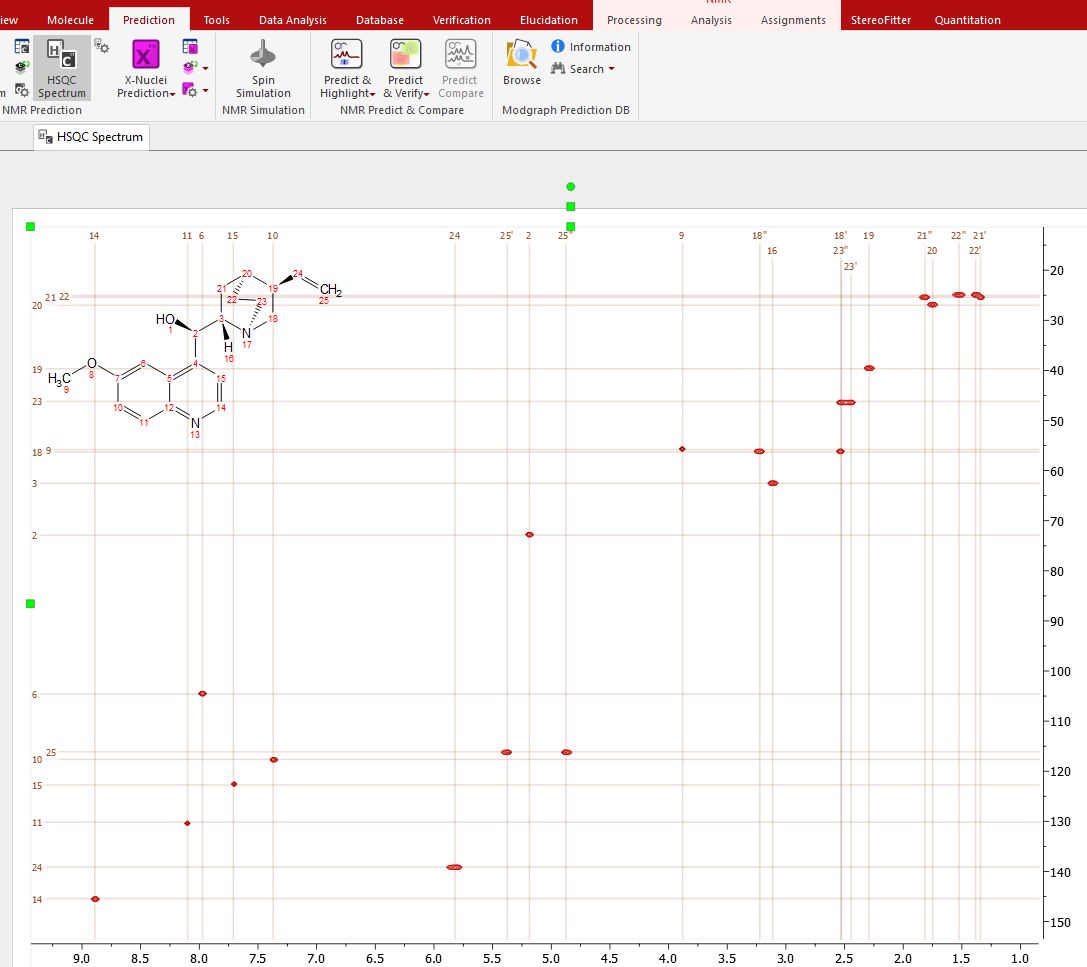
Follow the menu 'Prediction/HSQC Spectra' to predict a HSQC spectra from a given molecule:
You can modify the predicted chemical shifts either graphically, by clicking&dragging any prediction label, or by typing the new values in the prediction tables:
The same procedure will be followed to run X-Nuclei predictions 11B, 15N, 17O, 19F, 29Si, 31P (under the menu 'Prediction/X-Nuclei Predictions'):
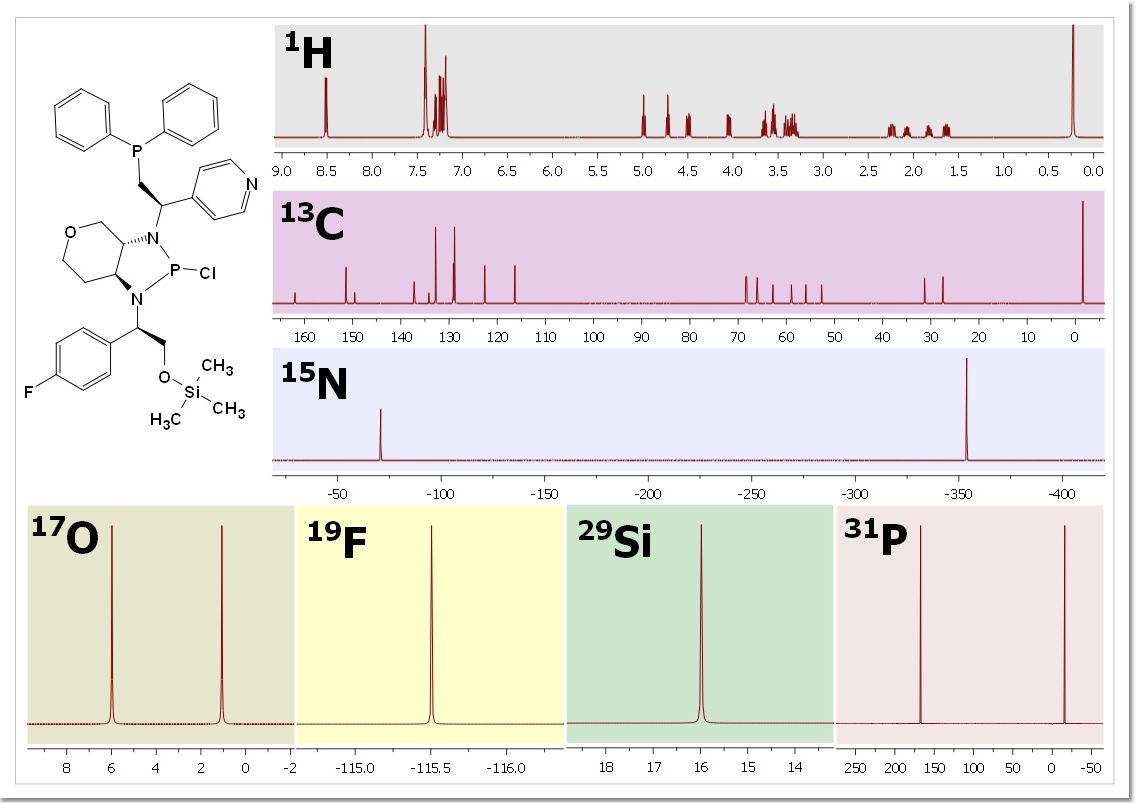
And here you can see an example:
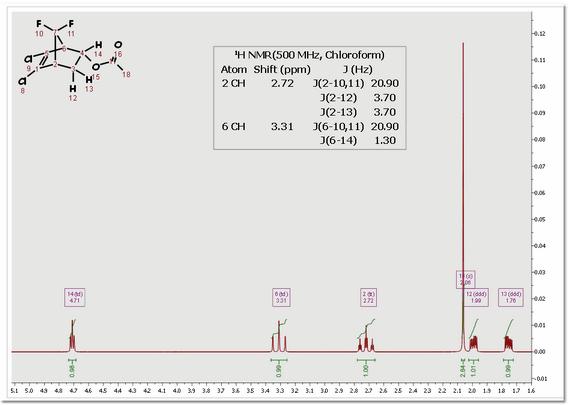
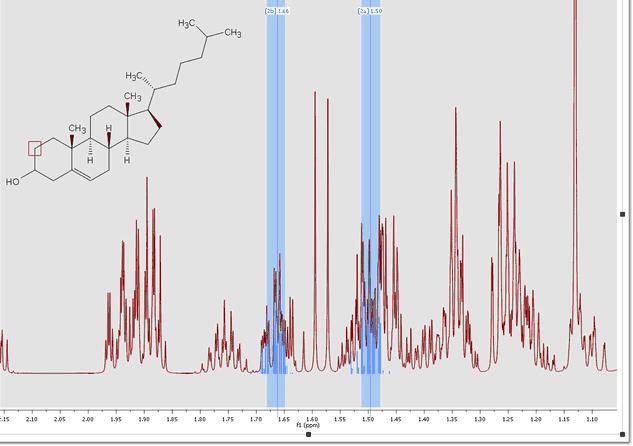
You can see (in the picture below) the higher order effects observed in the 1H predicted spectrum of the cholesterol:
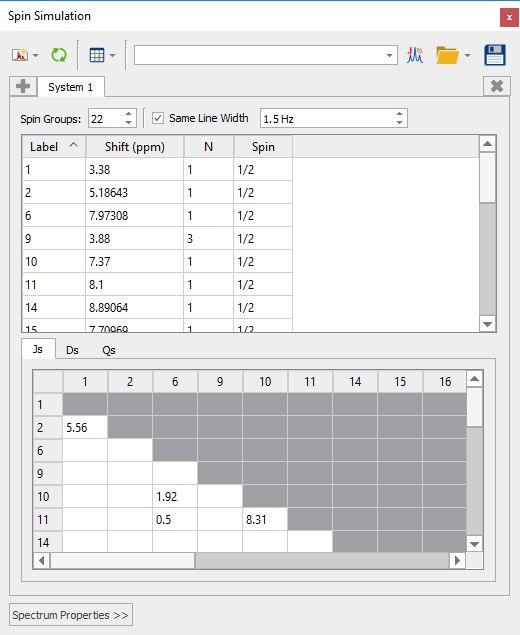
After having run an NMR prediction, the Spin Simulation panel will be automatically populated with the predicted values:
Modifications done in the spin simulation panel will be reflected in the 1H Prediction table (an vice versa); those changes will be graphically updated in the applicable synthetic spectrum. See also: |