Prediction Options and Properties



Prediction Options and Properties |
|
|
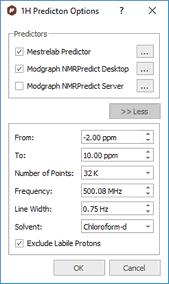
Following the menu Prediction/Prediction Nuclei Options will display a dialog box from where you will be able to select the chemical shift limits of the spectral window, as well as other parameters, such as number of points, frequency, Line Width, solvent and minimum coupling constant value. If you are going to predict a 1H-NMR spectrum, you can also exclude the 'Labile Protons' from this window
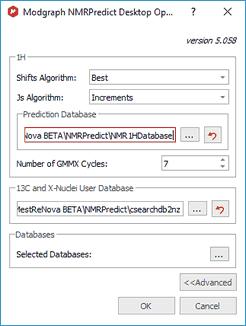
Clicking on the 'Predictor Properties' button will display a dialog box to allow you to select the location of the prediction Databases and also the algorithms to use (Best, Increments or Conformers) for the shifts and Js in the 1H prediction:
The 2D mol file representation of the structure is converted by GMMX into a 3D structure and then this structrure is repeatedly minimised to produce a series of conformers.The parameter "Number of GMMX Cycles" affect the number of conformers produced by GMMX. There is, however, no simple relationship between this value and the actual number of conformers.
Clicking on the 'Selected Database' will allow you to select the 'Prediction Databases' that you want to use for your predictions.
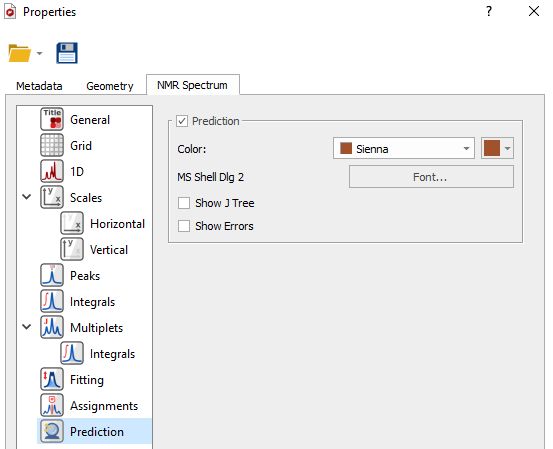
Properties Double clicking on the predicted spectrum will display the 'Properties' dialog box:
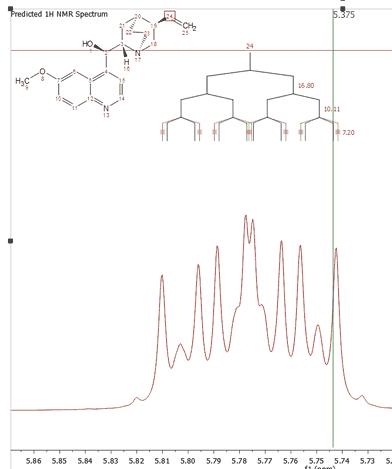
Selecting 'Prediction' from the left tabbed menu will allow you to change the color and font of the prediction labels and also to display graphically the J Tree and the prediction errors:
|