Preferences



Preferences |
|
|
Follow the menu File/Preferences/Mass to change your MS preferences:
The user will be allowed to select the saving options (item only or item+dataset).
You will find also an option to fit to height after zoom (selected by default). When this option is checked, a fit to height is applied after making a horizontal zoom in a mass spectrum or a chromatogram. In case of the other zoom modes (vertical or in both dimensions), a fit to height is never applied.
When the option 'Synchronized chromatogram's Retention time' is selected, you can zoom, pan and cut to all showing chromatograms. An analogous action for synchronization of the m/Z scale of mass spectra is selected by default. When checked, the "Synchronize Cromatograms RT" and "Synchronize Mass Spectra m/Z" buttons in Mass/Tools/Syncronize show enabled by default, for example when loading a raw Mass file. When unchecked, the "Synchronize" buttons in Mass/Tools/Syncronize show disabled, for example when loading a raw Mass file.
The "Live Mass Spectrum Preview" will allow you to show the extracted mass spectrum on the fly when you hover the crosshair over the chromatogram (or trace). After having clicked at the desired retention time, you will get the expected MS spectrum displayed.
The 'Correlated Crosshair' mode will display the crosshair as usual in the active plot and also as a semi-transparent crosshair in the none-active plots of the same type (chromatograms, mass spectra or UV spectra).
"Isotope Cluster Selection": After having extracted a MS spectrum if you click on a mass peak label, a zoom is automatically applied trying to show the isotope cluster. This zoom can be tuned by changing the values of the maximum distance (between the peaks forward and backward) and the intensity bigger ratio.
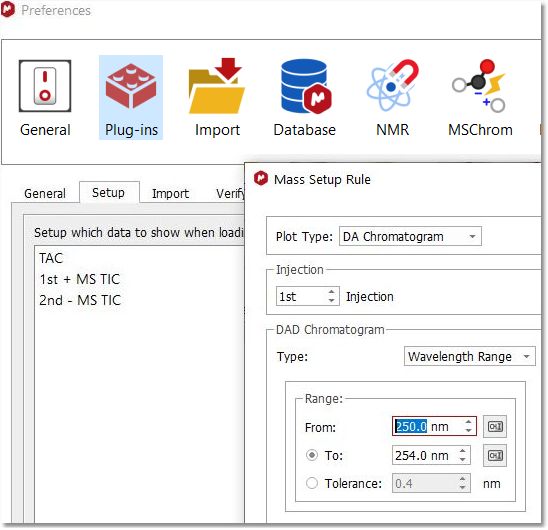
It is possible to choose which chromatogram and spectra are displayed when loading an MS dataset via File/Preferences/Mass/Setup. This mass setup will be stored when saving the preferences:
You can define rules to apply 'Traces Auto Alignment' or 'Custom Run Time ranges' automatically when importing a mass dataset:
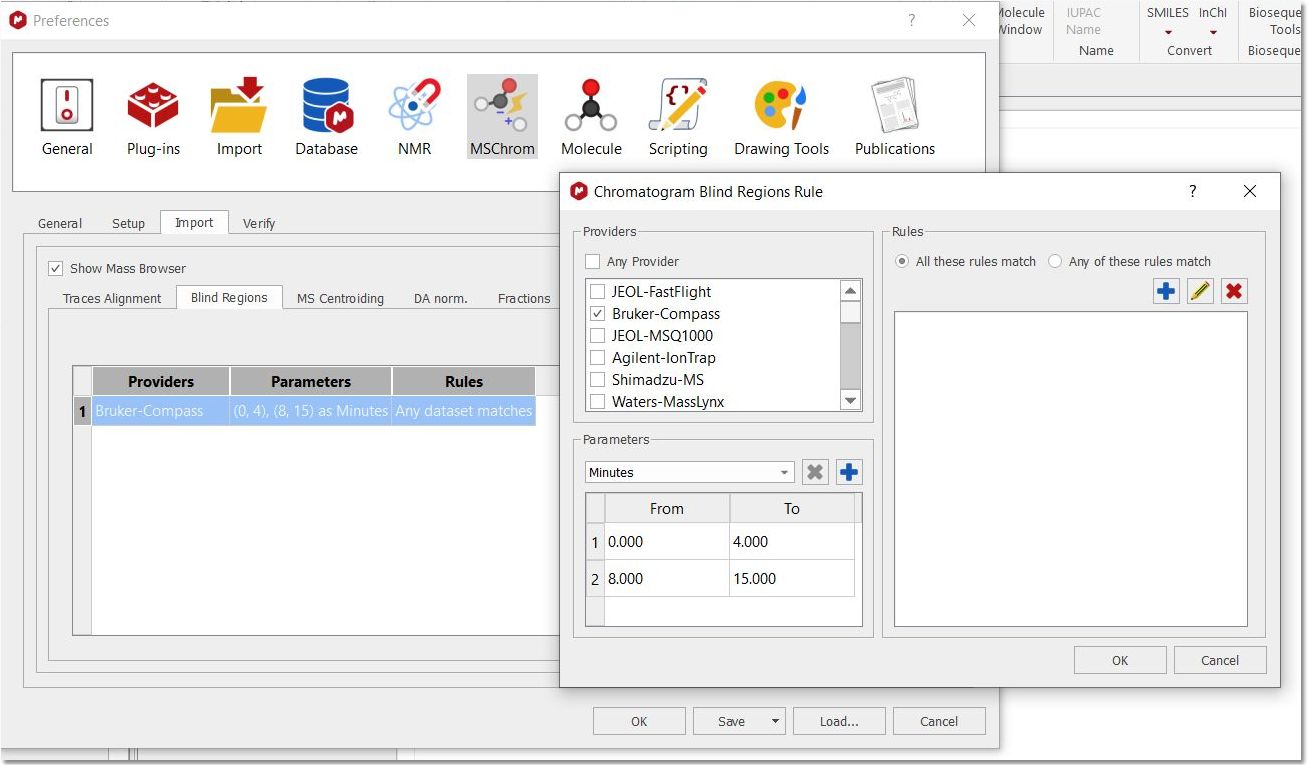
You can define default blind regions from the applicable tab:
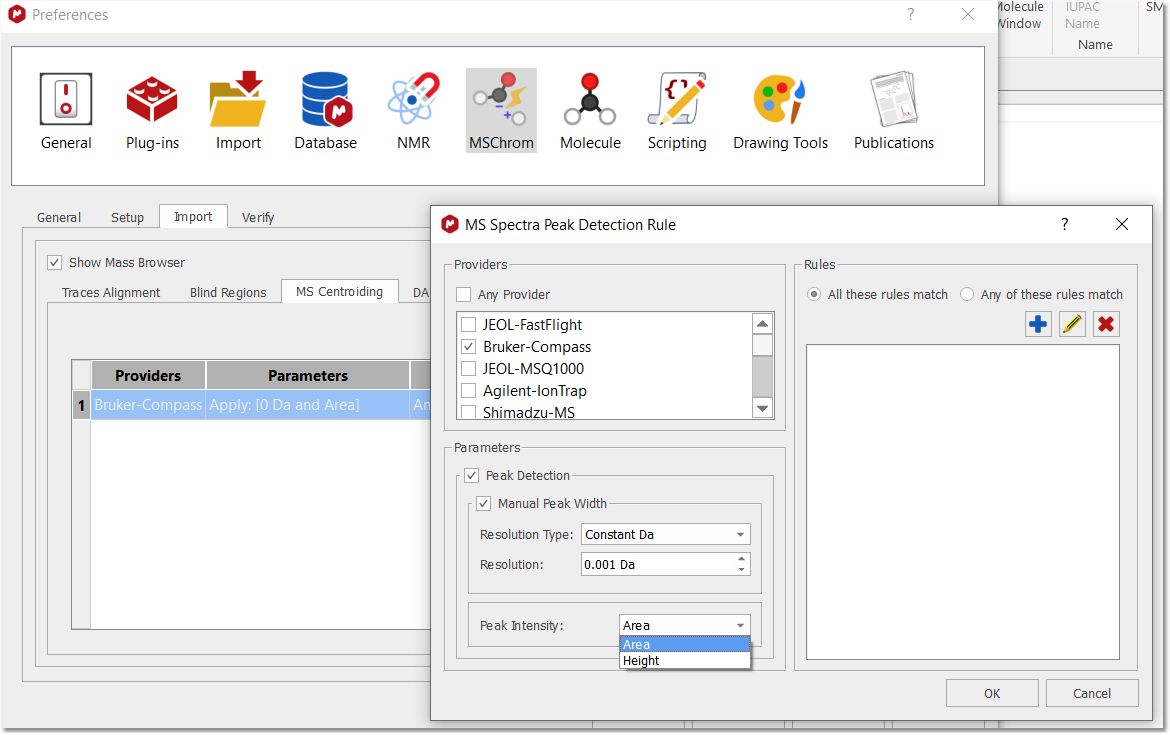
From the MS centroiding tab, you can add rules for peak detection, to be height or area based according to the selected provider (make sure that you restart Mnova after having applied the change):
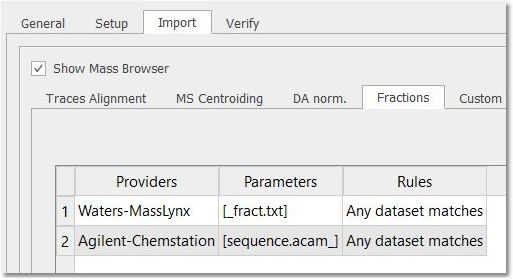
For Waters Masslynx datasets, you can highlight the fractions imported from the _fract.txt file. The same can be done for Agilent-ChemStation datasets, by importing the fractions from the sequence.acam_ file.
From the advanced tab, you will be able to customize the cache settings for your spectra collections:
Backing Store File: When enabled, all the spectra read from a dataset file, will be stored in a temporal file, so the next time we want to get the same spectrum, Mnova will get it from that file instead of reading it from the dataset by using the vendor libraries. This would benefit those providers using 32-bit libraries, like Advion and Varian. Memory cache: In addition to the backing store, you can keep the spectra in memory (which would have an impact in memory usage): oDisabled: No memory caching is done. oUnlimited: The spectra will be cached in memory without limitation, until you run out of memory. oLimited: The spectra will be cached in memory, but only the last spectra that do not exceed the set memory amount. |