Molecules Editor



Molecules Editor |
|
|
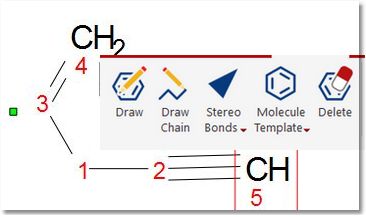
Mnova incorporates a basic 'Molecules Editor' which will be improved in the forthcoming versions of the software. This editor includes a powerful toolbar to draw your molecules:
The Drawing mode will allow you to draw a Carbon on your spectral window just by clicking on any empty field. Clicking on any existing atom will add an additional carbon. If you click on a single bond, you will get a double bond (clicking on a double bond will draw a triple bond).
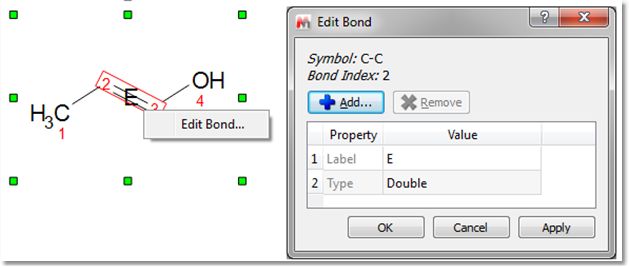
The same result can be obtained from the bond context menu (by right clicking on the bond). The 'Edit Bond' dialog will allow you to change the bond type (single, double, triple, up, down) or to set a label for the bond (to add stereochemistry notation, for example):
You can change the atom just by hovering the mouse over the atom and typing the applicable letter (i.e 'o' for Oxygen, 'n' for Nitrogen, "b" for Boron, 'b' plus 'r' for Bromine, or more complex labels, such as Me, Ph, COOH, etc). Make sure that you have in the 'File/Preferences/Molecule' the option to have the keyboard editing always available.
You can also change the atom by double clicking on it to display the 'Edit Atom' dialog. From here you will be able to change the name (from C to F, for example).
If you have any spectrum loaded together with a molecule; you will need to select any molecular drawing tool (from the molecular sketcher toolbar) before typing the applicable letter (unless you activate a 'Molecule preference" to have the "keyboard editing" always available). See more here.
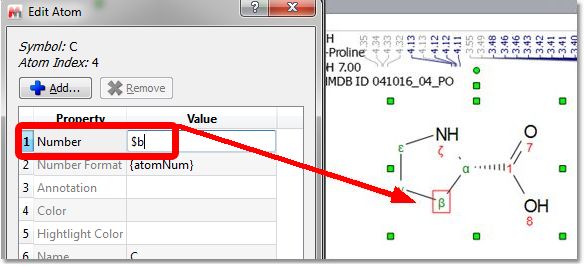
From the 'Edit Atom' dialog, you can add greek letters to the number by typing the $ prefix; for example $a, 4$b, 22$g$d21 will be converted into α, 4β, 22γδ21.
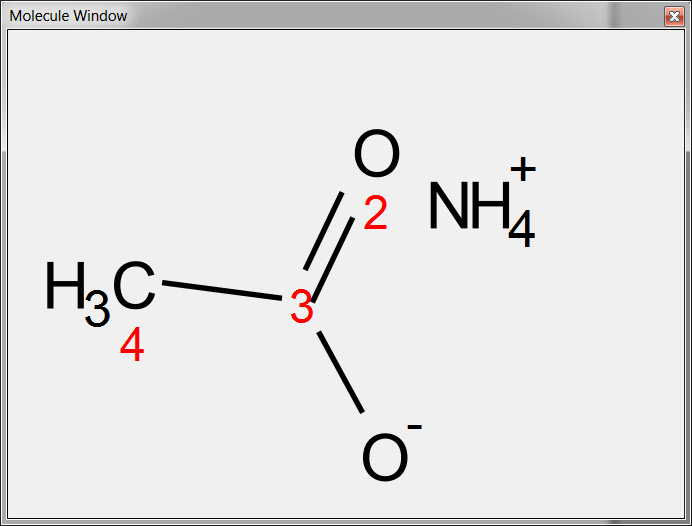
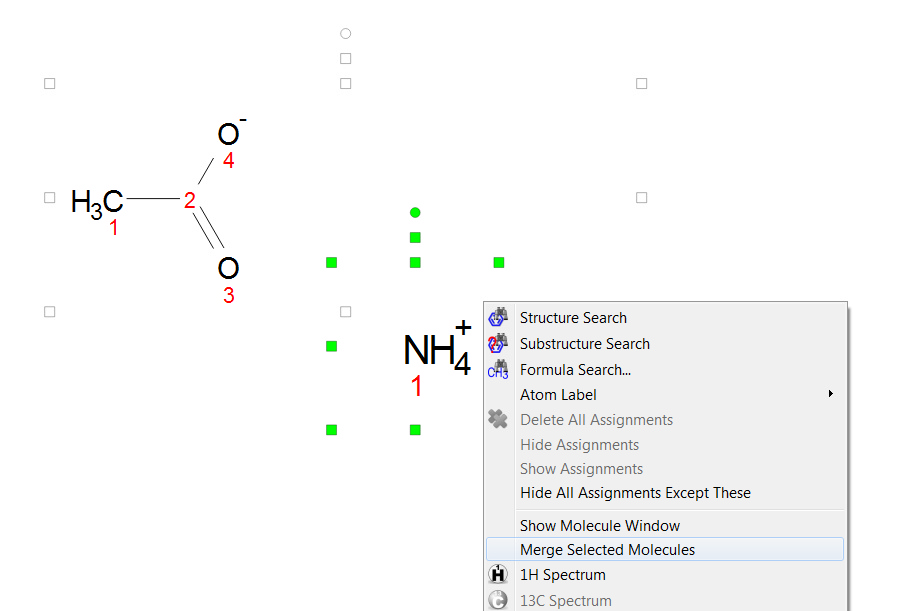
You can draw salts from the 'Molecular window' panel (View/Panels/Molecule Windows'), just by drawing both components there. To draw the NH4+ component, you will need to draw a CH4 and then hover the mouse to type the N followed by the + (to add the positive charge):
You will obtain the same result by drawing both components, selecting them, right clicking on any of the molecules and choosing 'Merge selected Molecules':
Mnova will not allow to add new atom (clicking on existing one) or change the bond type if that leads to atom valence exceeding, but it will allow you to create a new atom by stretching the bond from existing one. In this case the old atom will be marked with invalid valence cross:
Mnova will also automatically change the atom valence (e.g. 3-5-3 for Nitrogen) when adding/deleting new atoms or changing the bonds type.
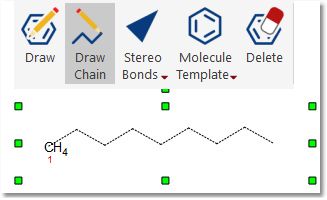
Draw Chains: draw chains by clicking, holding and dragging after having clicked on this button:
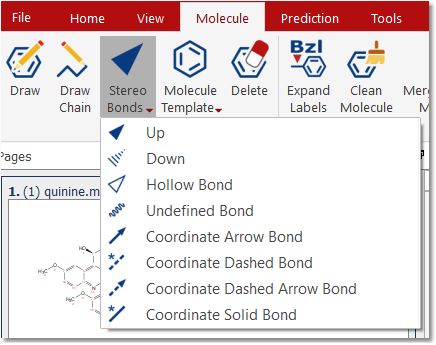
Stereo Bonds: Use this scroll down menu to draw stereo and coordinate bonds Clicking on any stereo bond will change its direction.
You can delete the bonds keeping the atoms (by holding down the Ctrl/Cmd key). Deleting several atoms selected with lasso while pressing Ctrl/Cmd key allows to keep neighbor terminal atoms.
The Molecule Template scroll down menu, will allow you to draw cycles:
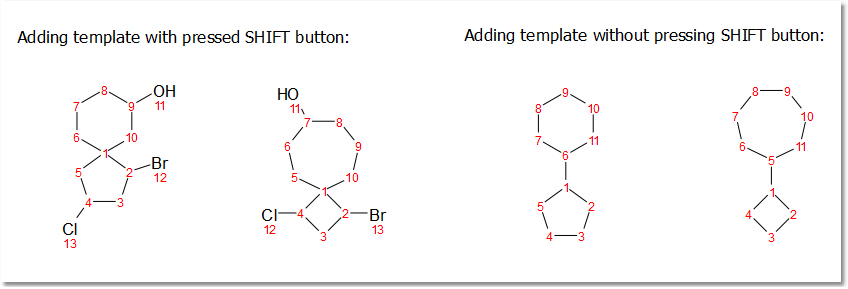
Spiro atoms can be drawn by adding cyclic templates while pressing 'Shift key'.
Clicking on the 'Molecular Templates' button will allow you load more templates. Once you have selected the desired template, you will only need to click on the molecule and click again on your spectral window to paste the molecular structure
You can add any template as spiro compound by holding down the shift key and clicking on the desired atom.
You can also create your own user templates by clicking on the 'Import' button and loading a .SDF file containing molecular structures or by clicking on the 'Add Molecule to user Templates' button of the 'Molecule Ribbon'.
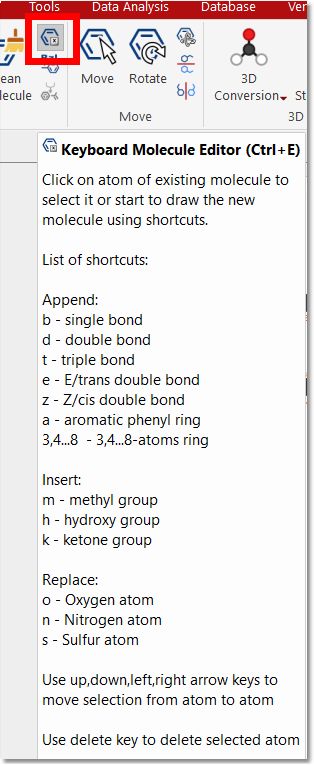
Keyboard Molecule Editor (shortcut: Ctrl+E): to draw your molecules without needing to use a mouse. You will find the shortcuts in the tooltip obtained by hovering the mouse over the button:
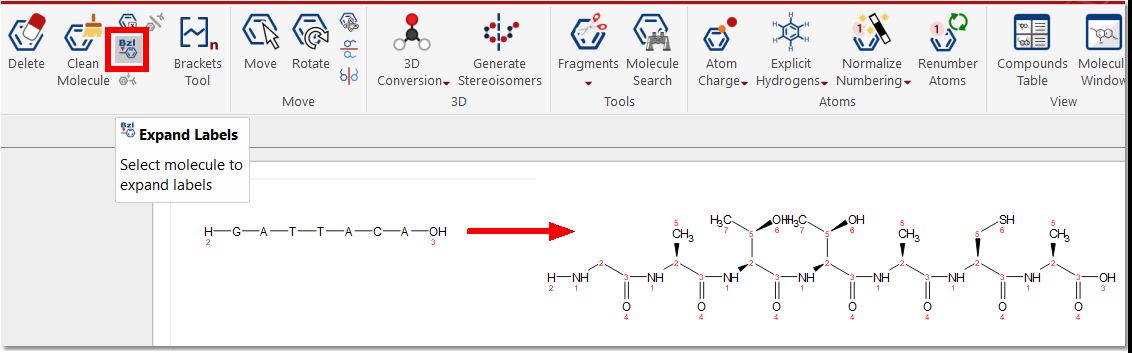
Expand Labels: to draw molecules with compacted groups and expand them by right clicking on the applicable group:
You can expand them by enabling the "Expand labels on importing" option in 'Preferences/Molecule' and also by clicking on the button "Expand Labels" in the Molecule toolbar.
Use the macro editor (under the General tab) to add free text labels and several parameters from the Molecule Properties.
The molecule label is stored in a layout template and also gets applied when applying the template. See also the Properties chapter (for further information about the macro editor)
Merge Selected Molecules: useful to merge several fragments into a unique structure.
Unmerge Molecules: to split a merged molecule into its individuals structures.
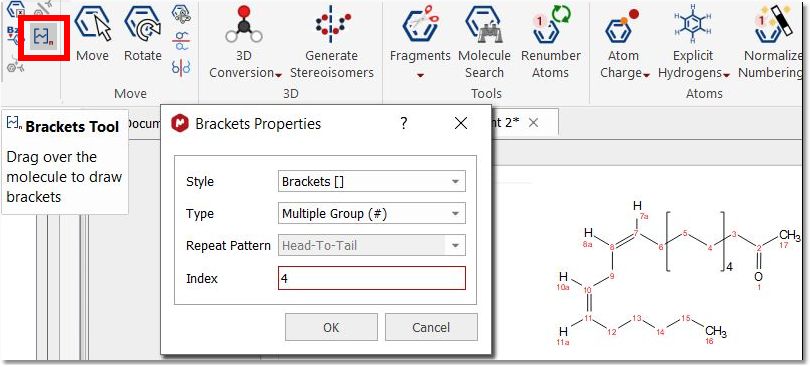
Brackets Tool: to draw polymers. Clicking on this button will display a dialog with different bracket options (style, type, repeat pattern and index).
The brackets can only be drawn while this dialog is opened. We have to select the desired properties and then draw the brackets over the desired region of the molecule. In this way you can draw as many brackets as you want (changing the settings if needed) before closing the dialog.
When drawing several brackets in the molecule, the last drawn is highlighted in red until the "Brackets Properties" dialog is closed or next pair of brackets is drawn:
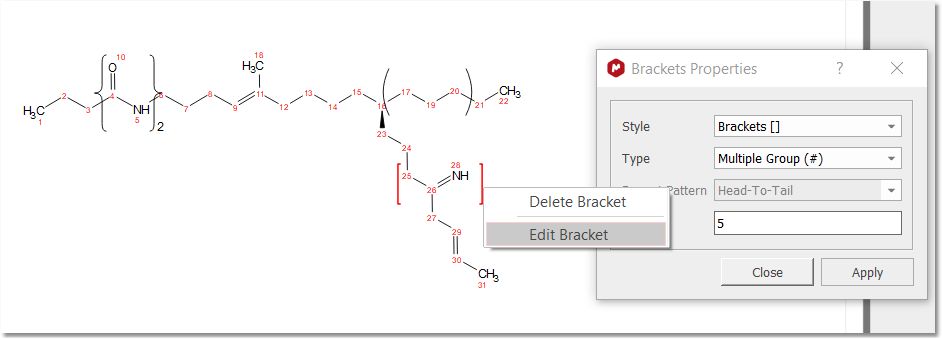
You can edit a selected bracket by right clicking on it to choose "Edit Bracket" from the context menu. The bracket will be highlighted in red and the "Brackets Properties" dialog will be displayed:
Move Mode: This mode will allow you to move an atom or a bond just by clicking&dragging.
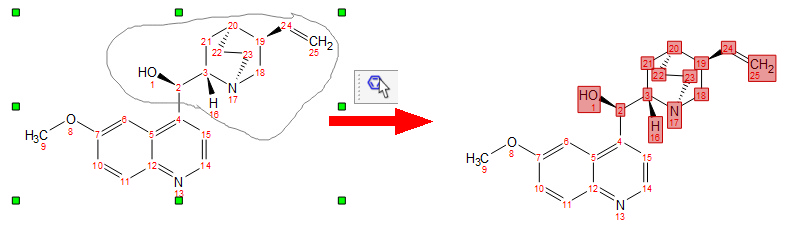
In this mode you can also use the lasso mode (by clicking and dragging the mouse around the desired part of the molecule) to select several atoms (to delete them, for example):
When you select an atom, move it over another one and release the mouse button, only the last atom will stay (except when the first selected atom is non-carbon and the second atom is carbon).
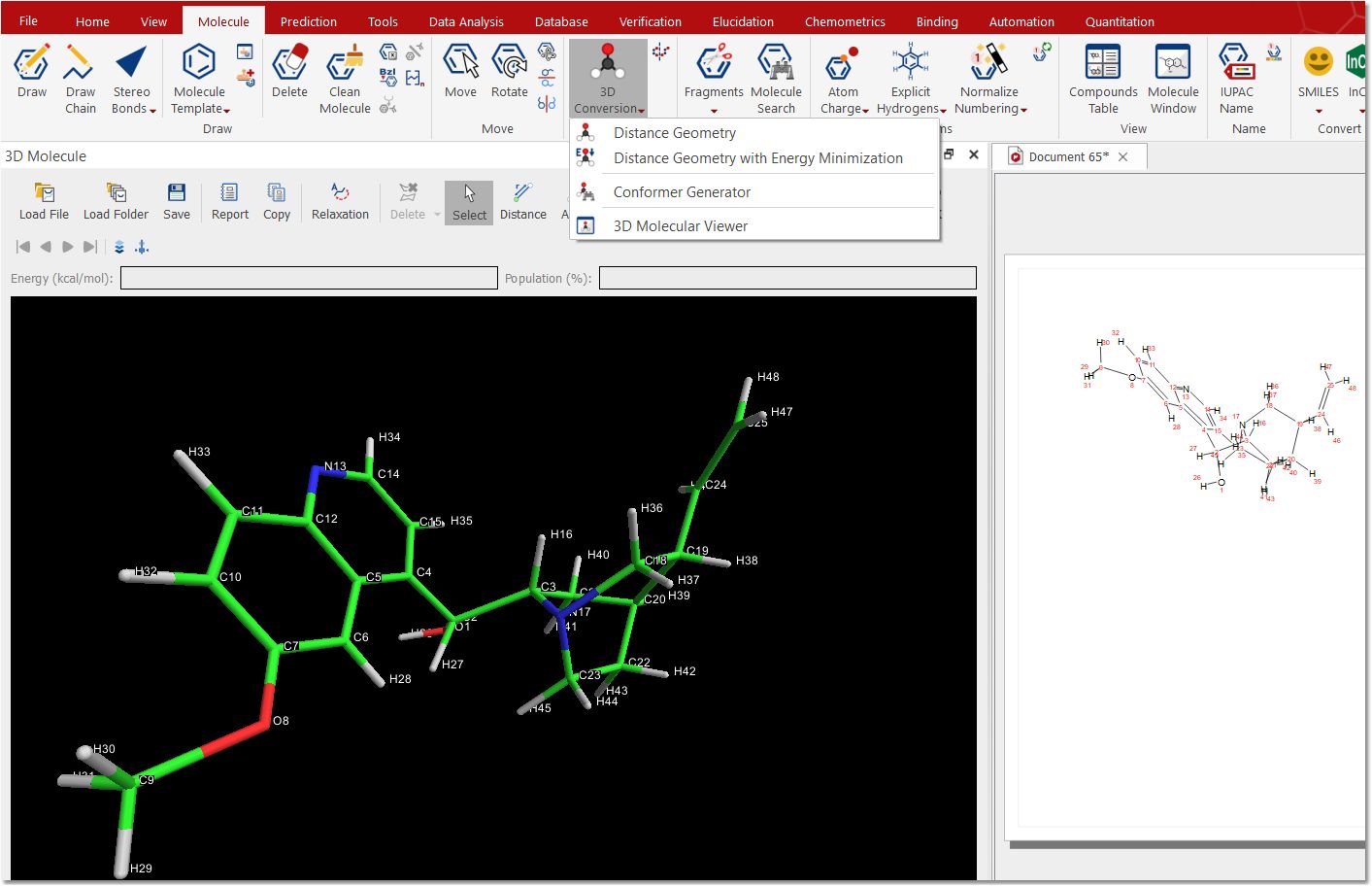
3D Conversion This feature will convert a 2D molecule in a 3D structure, calculating the energy for each conformer and their population.
NOTE: If you are not getting the 3D molecule in the viewer, please change the OpenGL engine from the 'Preferences/Interface' dialog.
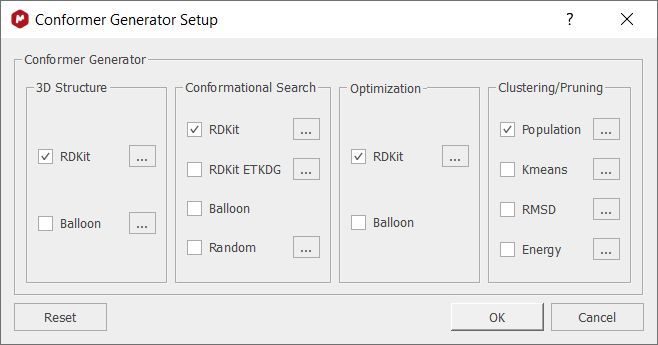
Clicking on the Conformer Generator option will allow you to select the algorithms for the calculation:
This allows the user to combine different engines, like gmmx, rdkit, and balloon to create list of 3D conformers. These lists can then be reduced via Kmeans clustering or RMSD, to produce a small list of the most relevant conformers.
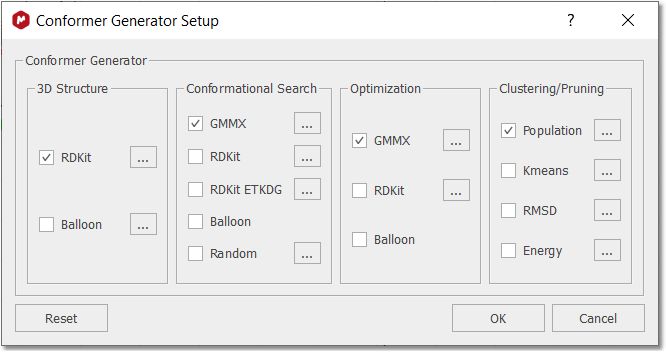
Users with a Stereofitter license will have GMMX conformation search and optimization capabilities:
The dialog is set out in four steps: Structure, Conformer, Optimization, and Clustering/Pruning. Each of these steps can be configured to run with different engines and each of these engines can be configured with different options. When clicking the Reset button, the default combination of engines and also the default options for each engine are retrieved
When loading a .mol file and using any of the available options, all possible conformers will be displayed in the 3D Molecule panel, including for each conformer: the Energy (kcal/mol) and Population (%).
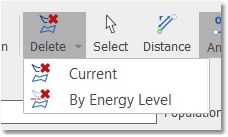
You could delete the undesired conformers just by clicking on 'Delete current conformer' button:
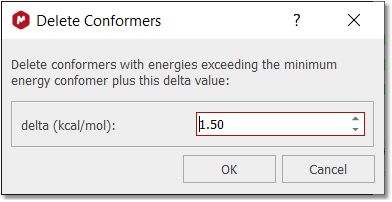
You can also delete conformers by energy level:
The '3D Conformation Search GMMX' tool will need a valid license for the Stereofitter plugin.
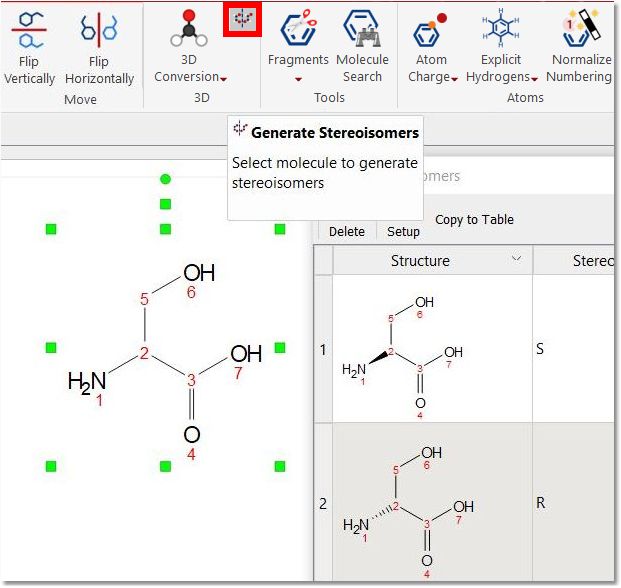
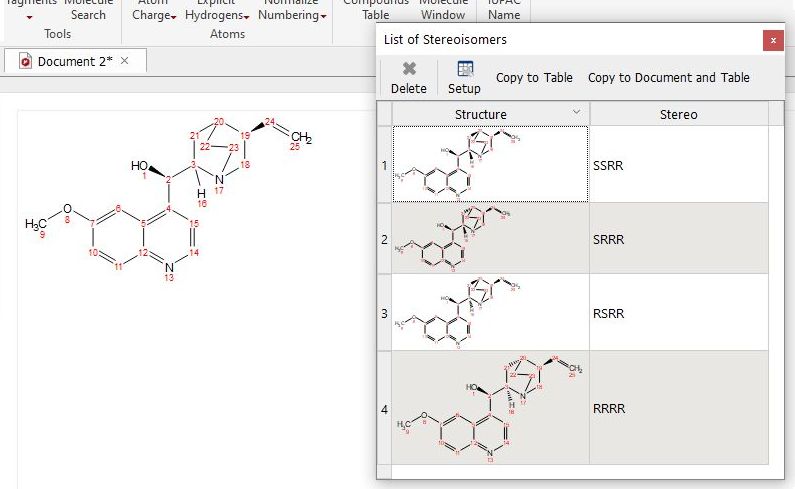
Generate Stereoisomers: it will display all possible stereoisomers for a given molecular structure (drawn with plain bonds).
If the given molecule contains any stereobond, that bond will be considered as fixed for the stereoisomers calculation.
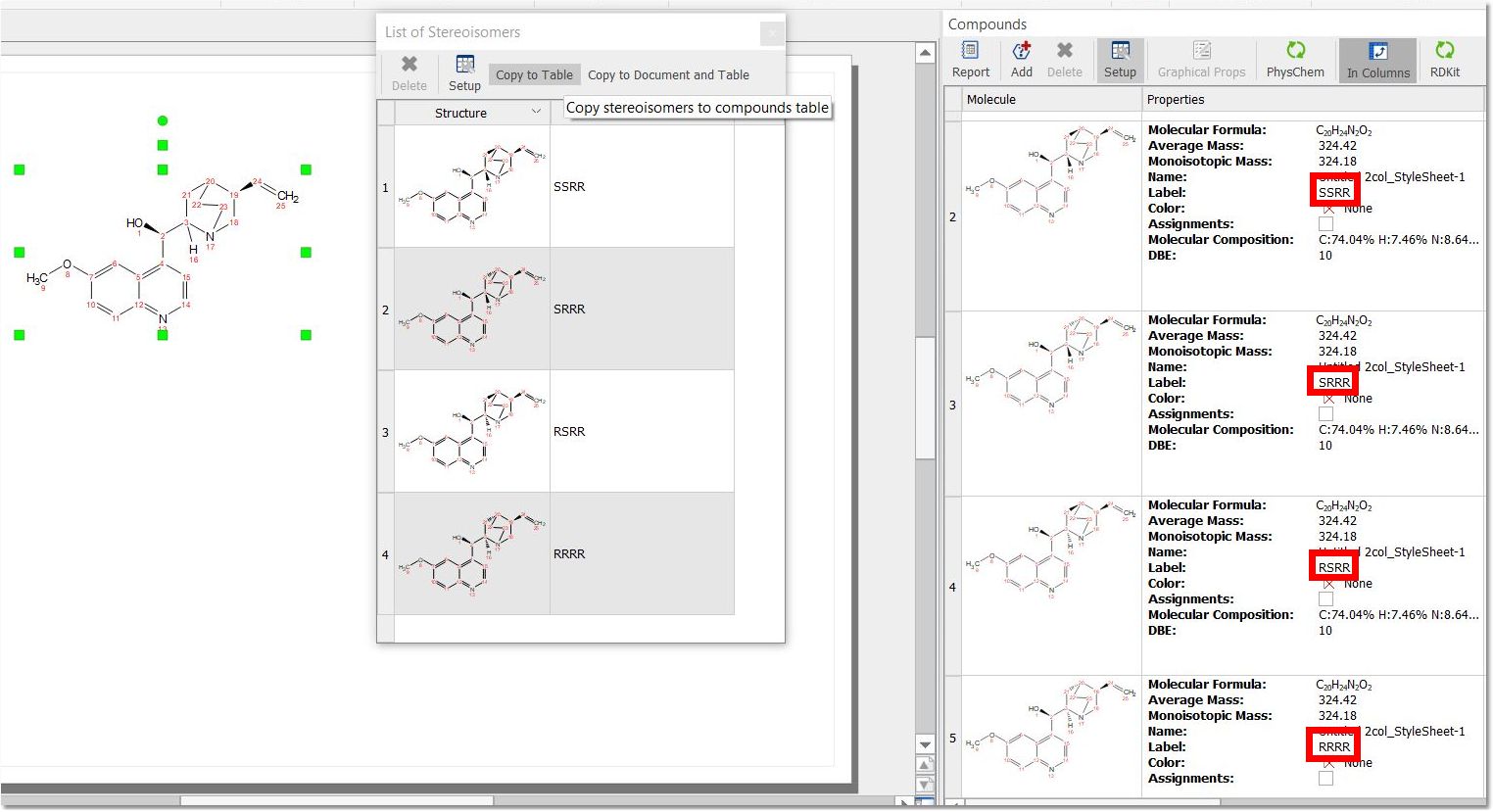
After having generated the stereosisomers, when copying them to the compounds table, the stereo info for each structure will be added in the "Label" tag:
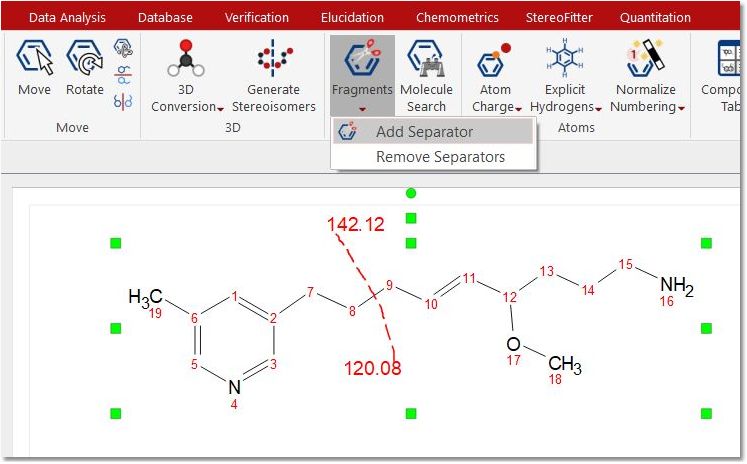
Tools: This ribbon will allow you to add/remove separators and to search for molecules in external Database (eMolecules and BMRB)
When selecting “Add Separator”, the mouse is changed to the icon of scissors and you can draw a tiny line in a structure (holding left button) to divide it. After that, a red dashed line is displayed over the molecule and the molecular mass of the fragments which divide the line are shown.
You can draw as many fragmentation lines as you want; all of them will be removed at once by selecting the “Remove Separator” tool.
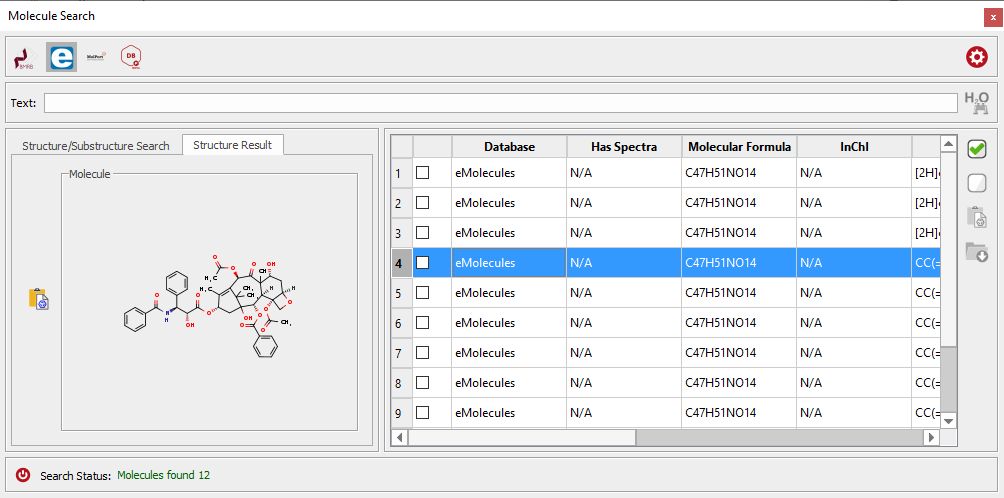
The 'Molecule Search' feature will allow you to search by molecular structure (or compound name) into the eMolecules and BMRB databases:
If a hit is found in the database, you can download the molecule and the spectra (if they are available) and work with them in Mnova.
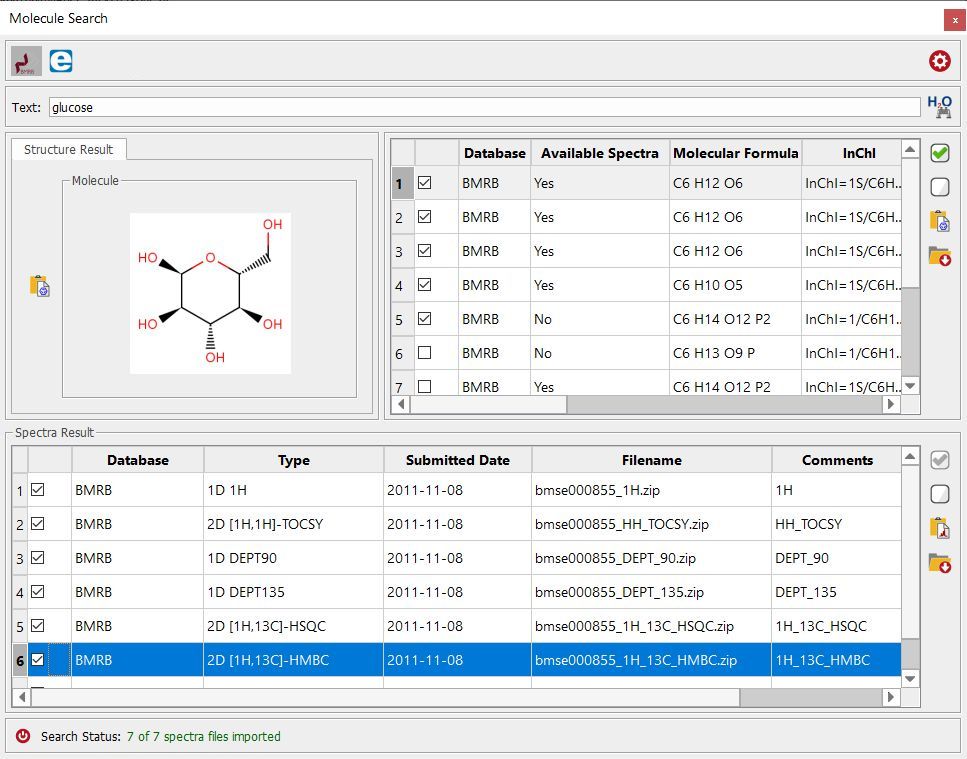
Here you can find an example of the glucose datasets found in the BMRB database:
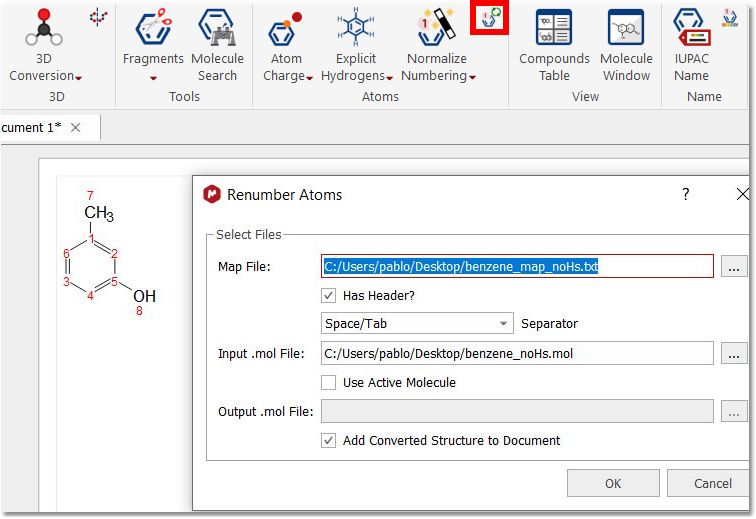
Renumber Atoms: it will take one map file, one input .mol, and one output .mol. The output will have the original .mol file but with all the atom numbers changed as defined in the map file.
The map file should contain two columns, the first one with the current numbering and the second with the desired one. Here you can see an example of a map file:
input output 1 1 2 2 3 5 4 4 5 3 6 6 7 7 8 8
Please bear in mind that you do not need a complete list of all of atoms, if you want to swap two atoms, you can use a map file like this:
input output 3 5 5 3
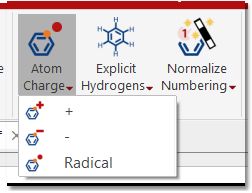
Atoms: This ribbon includes several features to add positive/negative charges (you can set a charge on any atom just by pressing + or - keys), radicals, add/remove explicit H and to normalize, lock or concatenate the numbering.
In the radical mode, after the 1st click on atom, the monoradical symbol will appear (· symbol). After the 2nd click, the diradical (Singlet) will be displayed (·· symbol). After the 3rd click, the diradical (Triplet) will appear (^^ symbol). Finally, after the 4th click, the radical will disappear.
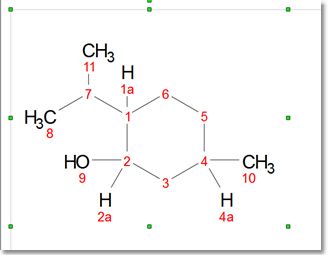
The Normalize Numbering button applies re-numbering of the whole selected molecule. From the Preferences you can select if you want to auto-renumber the imported (or pasted) mol files (and also when joining fragments); very useful if you have two equal molecules drawn in different applications or in different order, and you want to normalize the numbering.
The tool "Normalize Explicit Hydrogen Numbering" renumbers only the explicit Hs to match with the heavy atom number (with appended a, b, c... letters):
The Lock numbering tool will fix the numbering for the selected part of the structure (using lasso). The fixed atoms will have a yellow background:
Clicking the 'Concatenate Numbering' button, will de-duplicate the atom numbers of all the molecules in the document. The highlighted molecules will be renumbered so that the atom numbers are not repeated (leaving a gap from one molecule to another). The order in which molecules get renumbered will depend on the size of the molecule (the biggest molecule will keep its numbering).
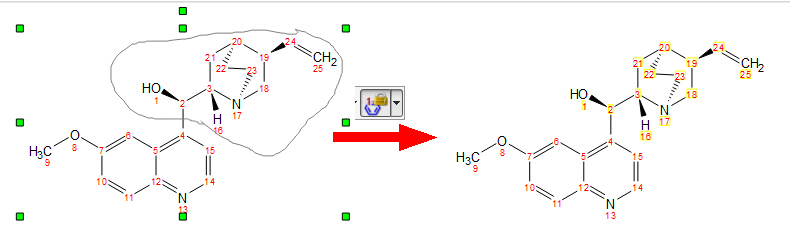
The 'Copy Numbering' feature, will takes the numbering of the selected molecule as template and applies it to all the molecules in the document which are equal to selected one. Very useful for the Stereofitter plugin when working with conformers with different numbering.
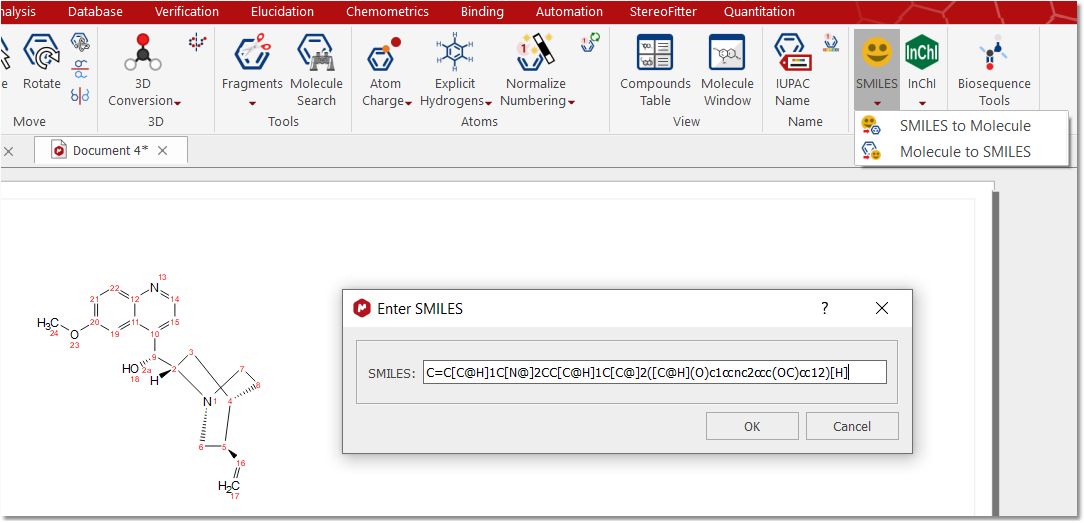
Smiles Mnova converts the smiles strings into the applicable molecular structure when you paste them from the clipboard or after having clicked on the "Smiles to Molecule" button:
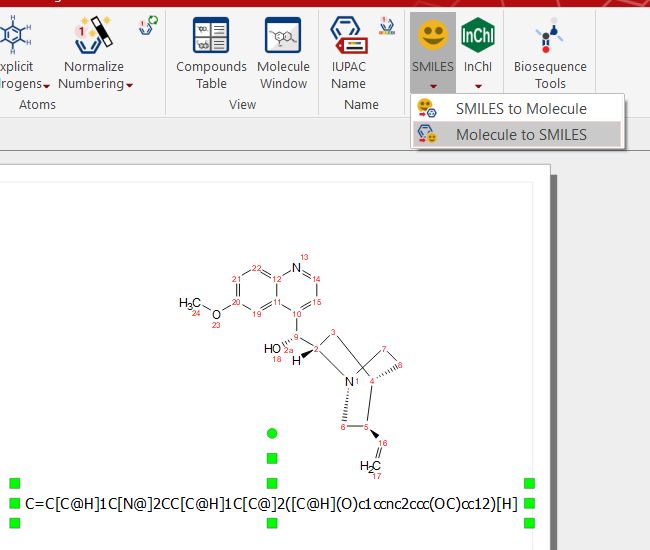
If you have a molecular structure, you can obtain the smile string by clicking on the 'Molecule to SMILES' button:
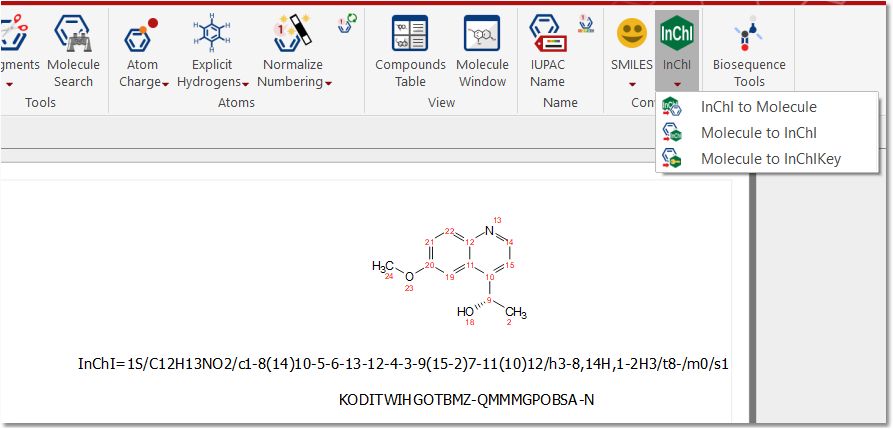
Inchi: You will have the capability to load molecular structure as Inchi, just by loading the applicable .inchi file into Mnova (or by clicking on the 'Inchi to molecule button' and pasting the Inchi string).
You can also convert molecular structures into Inchi and into Inchi Key functions.
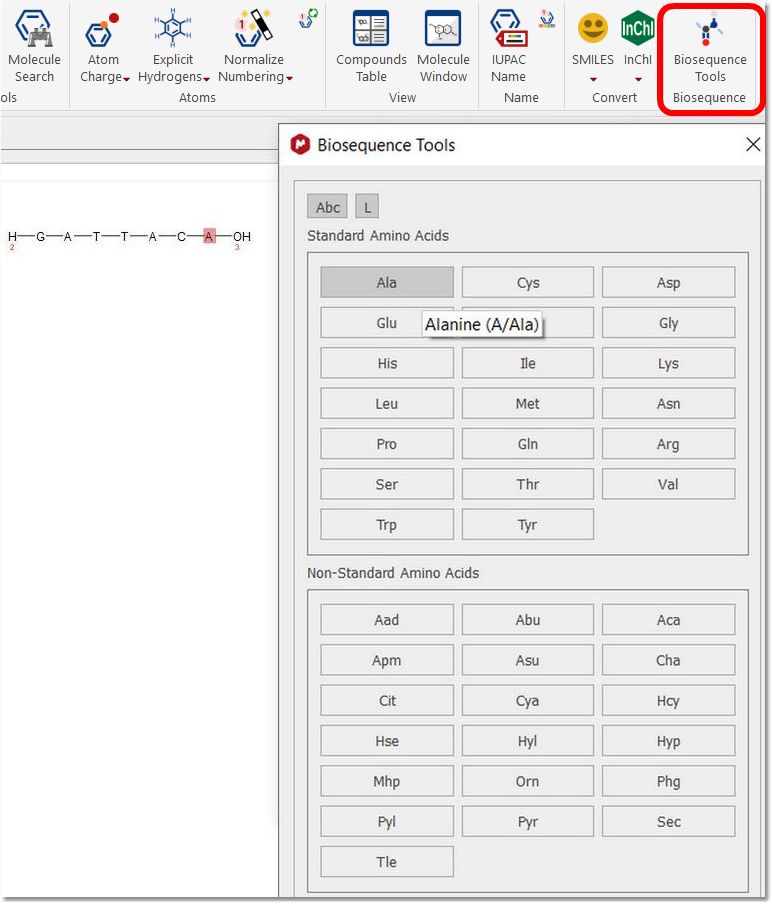
Biosequence: When clicking on the “Biosequence Tools” button, a dialogue will be displayed to allow you to draw peptides using the 20 standard amino acids and several non-standard:
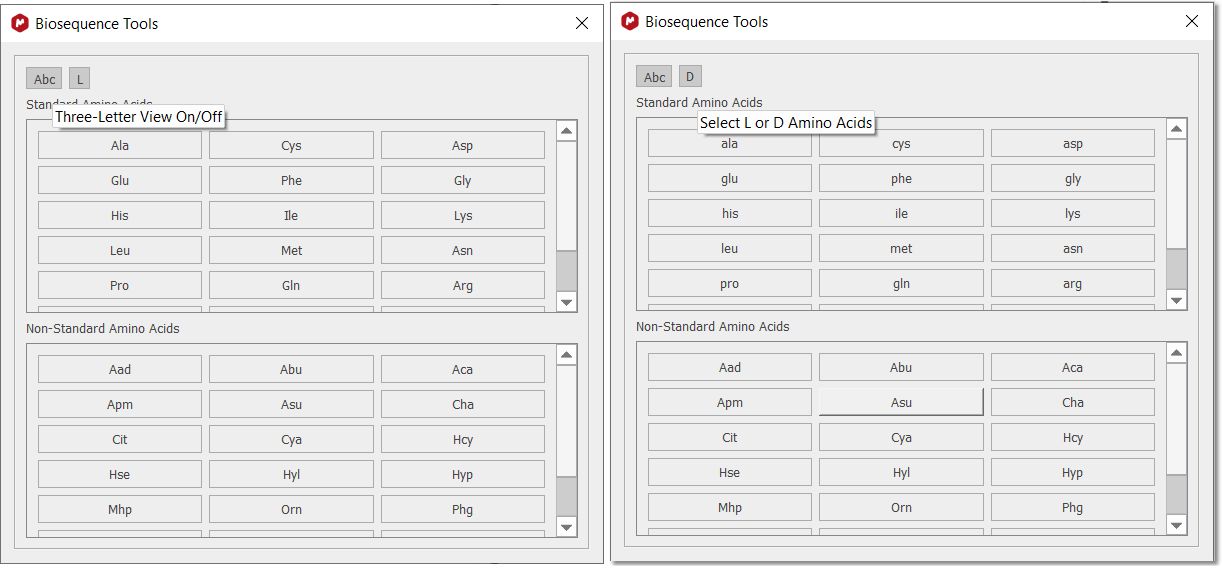
Once the new “Biosequence Tools” dialogue is opened, the cursor is changed to a peptide mode and only peptides can be drawn by using the 1 or 3 letters code. You can add D or L aminoacids, just by checking the applicable toggle button:
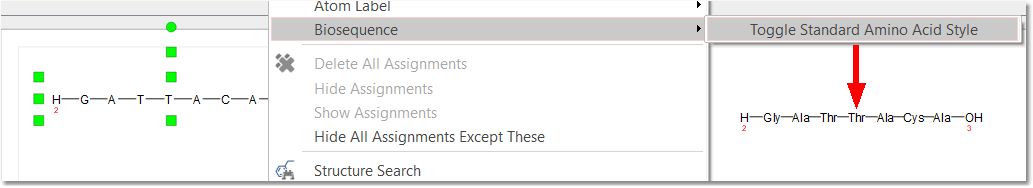
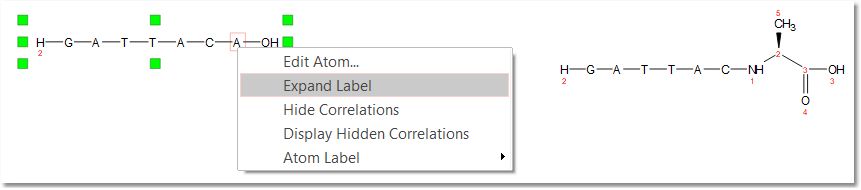
Once you have drawn the peptide chain, you can toggle the sequence by right clicking and selecting the applicable option from the context menu:
You can add amino acides inside the chain by selecting the AA in the chain and clicking on the desire AA that you want to add. To delete AAs just select the "Delete Atom/Bond" mode from the molecule ribbon and click on the AA (hovering the mouse over the AA and pressing the Delete key will also work).
Once you have drawn the peptide chain, you could expand the amino acid just by right clicking on it and selecting 'Expand Labels':
You can expand the entire peptide sequence by clicking on the “Expand Labels” button from the molecule ribbon:
|