Reference Alignment



Reference Alignment |
|
|
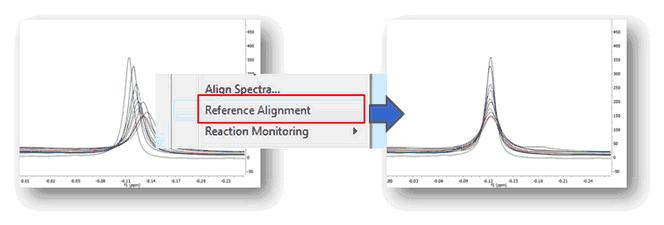
This feature will be used to manually align the chemical shift of 1D or 2D spectra (very helpful in Metabonomics), that is, to correlate signals of 1D or 2D spectra. Just select, in the page navigator, the spectra that you want to align and follow the menu 'Stacked/Reference Alignment'. All the spectra will be aligned with the first one.
You can also use this feature to align stacked spectra. In that case, the method uses the active one and then aligns all remaining spectra by maximizing the cross-correlation between the reference spectrum and the spectra to be aligned:
The idea of global alignment is very simple and corresponds to the well-known chemical shift referencing method in which the user sets the internal reference peak (e.g. TMS, DSS, TSP, etc) of each spectrum to e.g. 0 ppm. In order to cope with small fluctuations in chemical shifts, this method seeks for the highest peak within a narrow (user-defined, auto-tuning option in Mnova) interval, as depicted in the figure above.
See also Align Spectra. |