Settings



Settings |
|
|
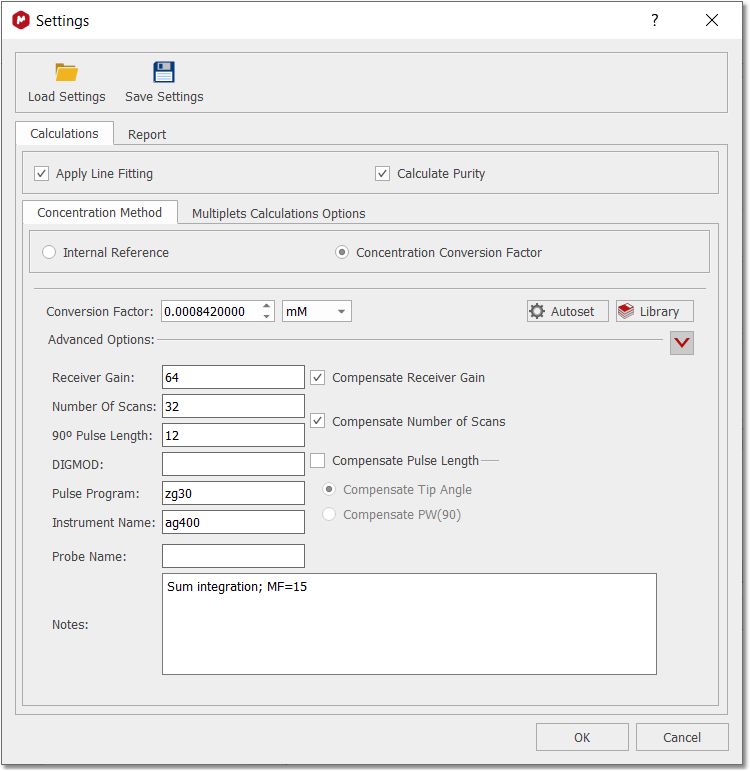
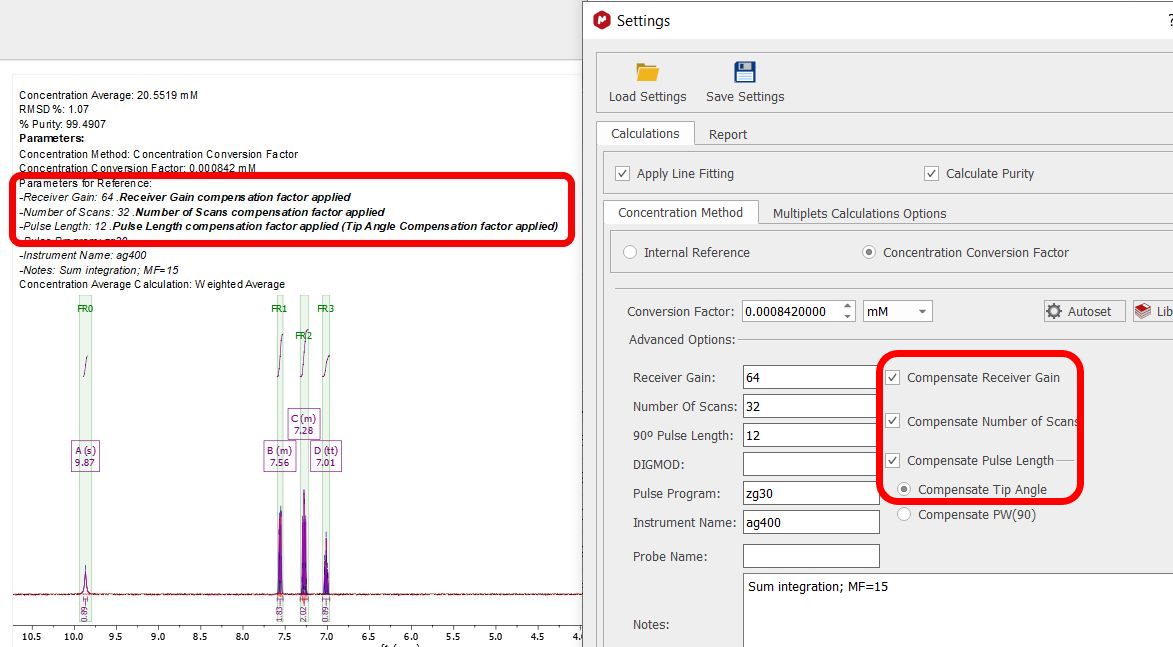
Selecting 'Settings' on the qNMR panel, will display the dialog box below, containing different tabs (Concentration, Options and Report):
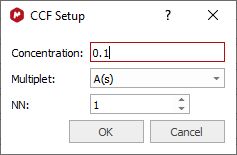
Note first the “Save” and “Load” buttons. These will allow you to set up the correct settings for parameters and distribute amongst other users to ensure they do the analysis correctly Please bear in mind that qNMR will use the integration calculation method that you have selected under the 'Multiplet Analysis' options. Calculations Apply Line Fitting. To apply the fitting over all the multiplets. Calculate area purity. If checked, this will show with the results summary. It is calculated using the sum of areas for compound and impurities (from GSD), and is something of a rough value: % Purity = (-A(C) / (A(C) + A(I)) * 100.0 A(C) is the sum of the GSD areas for peaks labelled as compound A(I) is the sum of the GSD areas for peaks labelled as impurity Concentration method Fundamentally, there are two ways to determine concentration: using a signal as an “internal reference”, or a specified “Concentration Conversion Factor” (CCF). The internal reference may be a real signal area (e.g., residual solvent, TMS) or an artificial signal (e.g. ERETIC or QUANTAS). If a residual solvent is used then the “Reference peak is a solvent” box should be checked, and then only peaks of type “Solvent” in the chemical shift range will be used for area calculation. This can be useful to exclude errors that would result if impurity, analyte or other peaks were used, and this is a nice feature that follows from AutoClassify. Concentration Conversion Factor Currently, we only support CCFs that have been determined using data processed within Mnova – there can be significant errors if Bruker 1r files are used. The CCF is determined for a particular experimental setup by processing the spectrum of a compound of known and specified concentration, and adjusting the “Conversion Factor” until the correct concentration is reported. To calculate the CCF value, just load the spectrum with the multiplet analysis done and click on the 'autoset' button of the 'concentration settings' panel to input the concentration reference, the NN for this multiplet and the applicable multiplet in the spectrum:
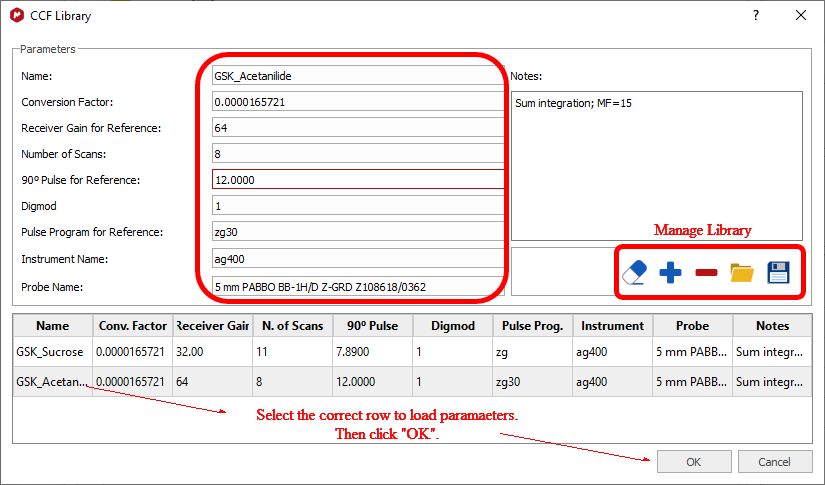
The CCF will be calculated using: CCF = [Reference]*(NN/AI) For further information about how to calculate CCFs with Mnova qNMR please take a look at this document. Library of CCFs It is important to account for the fact that a CCF value will have a fundamental, strong dependence on the spectrometer hardware: the particular NMR spectrometer and probe, principally. This must be determined each time empirically, as described above. After that is done, simple mathematical compensations can be made for differences in the spectrum acquisition parameters: receiver gain, number of scans, or 90° pulse length. If necessary, a different value can also be determined and saved for different solvents. To facilitate this, the user can save these values to a file – and distribute them, of course.
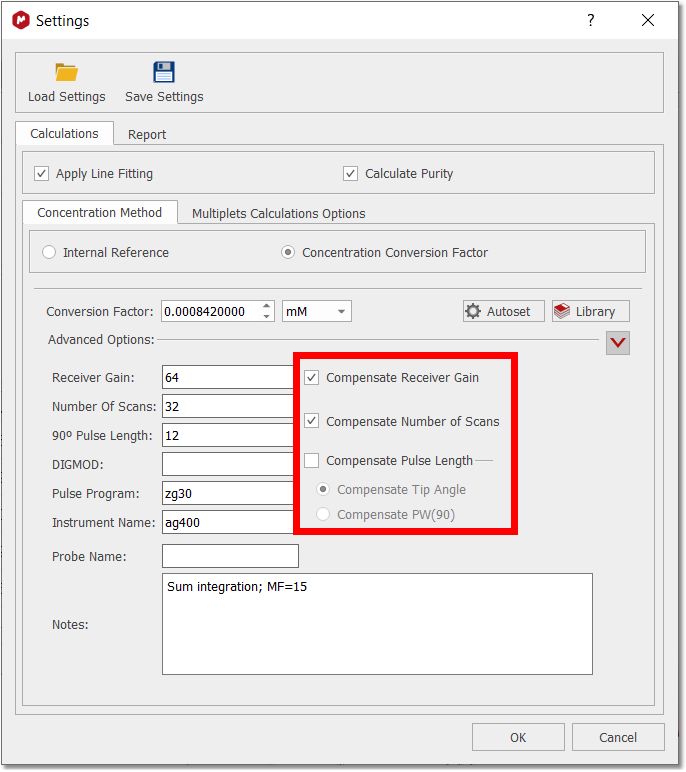
To add a new library condition the user will populate the parameter fields correctly, click the blue “+” button to add to the library, and save the updated data file. To select a different CFF, highlight the row and click “OK”. The software will check that the parameters match if specified, and this information will be included in all reporting. It is intended that the sample and references are acquired under same conditions, but you can compensate the receiver gain, number of scans and pulse lenght by checking the applicable radio buttons:
Here you can find how the compensations are applied and calculated: Compesated Integral = Absolute integral * Factor. •Factor to compensate Receiver Gain:
Factor = RG reference / RG experiment
Factor = [10 RG reference - RG experiment] 1/20
•Factor to compensate Number of Scans: Factor = NS reference / NS experiment
•Factor to compensate Tip Angle: Factor = PW reference / PW experiment
•Factor to compensate PW: Factor = PW experiment / PW reference
The concentration report will include information about the compensation factors applied in the analysis.
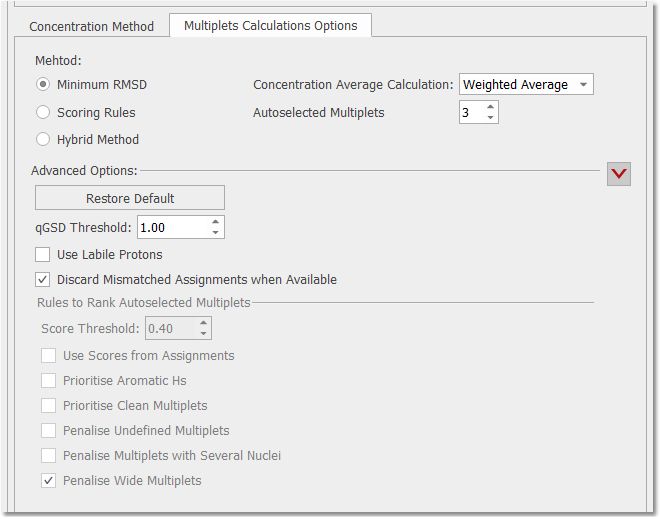
Multiplets Calculation Options This is where the settings for the multiplet selection criteria are grouped:
These methods will all try to select the number of multiplets in, whilst respecting the score threshold. Minimum RMSD. This option will ignore the multiplet scores and simply chose the multiplets that give the smallest RMSD in the average concentration. It may be used for very pure compound spectra. The maximum number to be used is the user-selectable “Autoselected Multiplets”. Scoring rules. This applies the rules selected in section 3, below, and then uses the top multiplets to determine the average concentration (and RMSD). The maximum number to be used is the user-selectable “Autoselected Multiplets”. Hybrid method. This method first applies the scoring rules and then chooses amongst the highest scoring multiplets. Using the multiplets whose final score exceeds the score threshold, the multiplets that give the smallest RMSD are identified and used for the final average concentration calculation (and RMSD). Concentration average calculation. Difference in how the average is calculated. Simple average will calculate the mean average of the concentrations. Weighted Average will calculate the concentration with the sum of the integrals, divided by the sum of the nuclei and multiplied by the scaling factor. Autoselected multiplets. The target (max.) number of multiplets that will be used for the final concentration determination. The score threshold, labiles use, etc. will be respected.
Advanced Options Use labile protons. The default is to ignore labile protons (if categorised), but if you check this box then the restriction is lifted. The thinking is that signals from labile protons almost always under-integrate, and should not be included in qNMR calculations. This offers a partial workaround if the user really wants to use labiles. This rule is applied if “Use Labile Protons” is checked; multiplets considered to be or contain labile protons will be penalized by a factor of 0.25. If unchecked, labiles have a score of 0.0 and are therefore omitted from all concentration calculations. Discard mismatched assignments when available. This test uses nuclide information from AutoAssignment and checks that it matched with NN from the multiplets. If they are not in disagreement, the multiplet will be discarded.
Rules to rank autoselected multiplets a.Prioritise aromatic Hs. Non-aromatic multiplets will have their scores penalised. Especially useful when the aliphatic region is complicated by large residual/impurity peaks or when solvent saturation is used. If the user checks the option, the aromatic Factor will be 1 for an Aromatic multiplet, and a non-Aromatic H is multiplied by 0.5 (Aromatic range=5.5-12ppm). b.Penalise multiplets with several nuclei. Prioritises multiplets having the fewest NN. This assumes that impurities can lead to a false NN. The score is multiplied by 1/NN: the larger NN, the lower the score. c.Use scores from assignments. If Verify/AutoAssign was used, the individual multiplet scores will be taken into account. A multiplet with a higher assignment score Q(-1 to 1) will be scored higher. Assignments Factor = 0.75 + Q/4 d. e.Penalise undefined multiplets. The thought is that a multiplet marked as “m” may be a clear solute multiplet but complicated by impurity peaks. In this case the “m” is penalised. The factor will be 0.6 for ‘m’ or 's' multiplet types, and 1 for all others. f.Prioritise clean multiplets. The presence of impurity peaks will cause a 10X increase in their relevance when determining the score. Use when spectra are very clean.
Clean Multiplets factor= multipletPeaksSum/(multipletPeaksSum+noMultipletPeaksSum)
multipletPeaksSum is the total sum of the areas of all the peaks noMultpletPeaksSum is the sum of the areas of the peaks not assigned to the multiplet There is a further correction for peaks labeled as impurities: the area for this peak type will be increased x10.
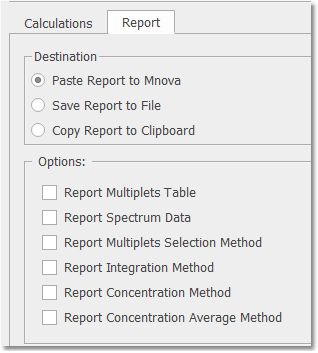
g.Penalise Wide multiplet. Penalises a multiplet having a width >60 Hz. This case can arise when the multiplet picks up impurity peaks. Consider using with dirtier spectra. The score is divided by 2 if the width of the multiplet is greater than 60Hz. h.Score Threshold. Multiplets having a score below this number will not be used in the average concentration determination. A value of 0.3 is typical. (Scores are between 1 and 0.). Reports This is where you can select how to report your results; by pasting the report into the Mnova document, by exporting to an ASCII file or by copying to the clipboard.
You will find also some options to choose which parameters you want to report. |