Line Fitting (Deconvolution)



Line Fitting (Deconvolution) |
|
|
When the spectrum is crowded with peaks, or affected by excessive noise, deconvolution may be the only way to measure the compounds ratio; but please bear in mid that this feature does not make miracles and it can not be a substitute for higher magnetic fields, more concentrated samples or longer acquisitions. The name “deconvolution” means (more or less): “removing the shape”. The theoretical shape of an NMR peak is given by the Lorentzian equation. Field inhomogeneity and weighting functions can however yield partial Gaussian line-shapes. From a merely graphical (and practical) point of view, they are two complementary shapes, in the sense that most symmetrical peaks can be approximated with suitable combinations of Lorentzian and Gaussian components. For your convenience, Mnova defines a mixed function, for which you can vary at will the percentages of Lorentzian and Gaussian character. MestReNova includes a fully interactive interface for the deconvolution of experimental spectra into individual lines.
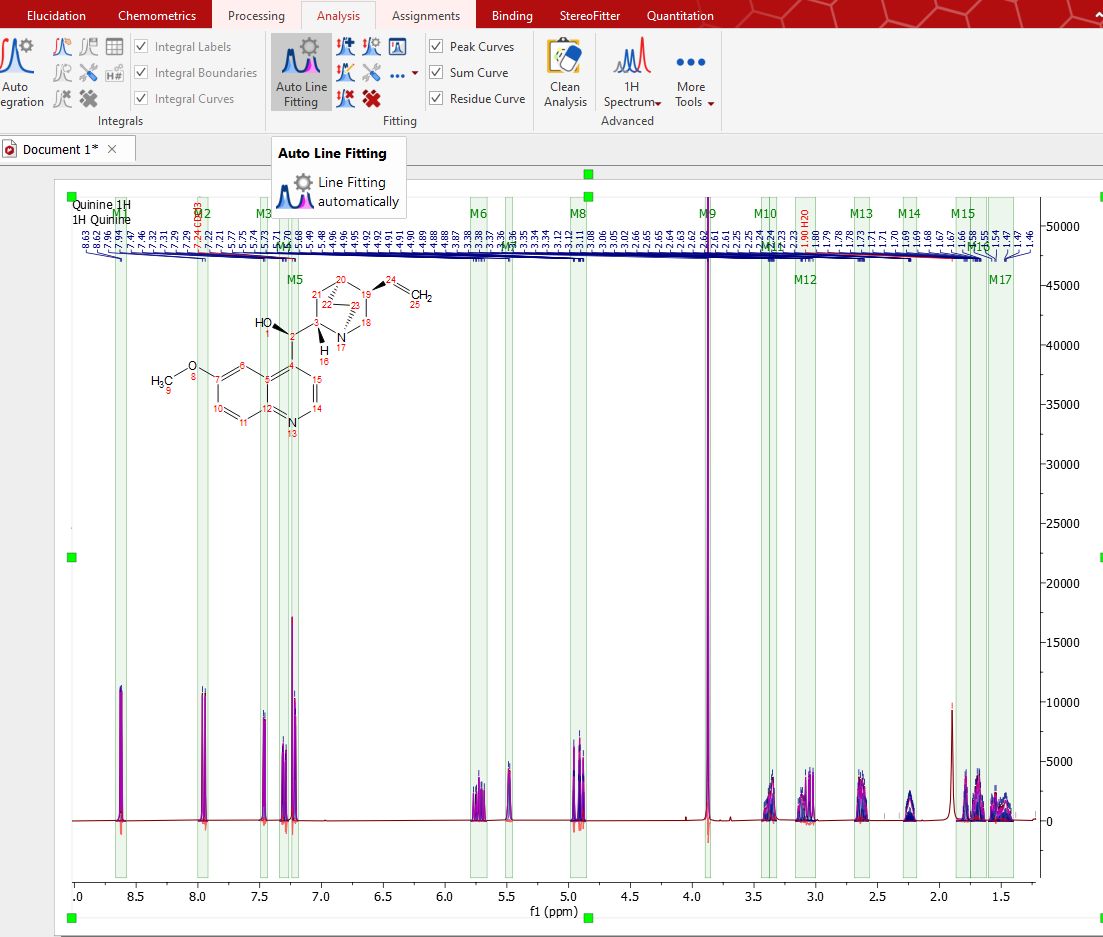
This module includes options for locking any of the parameters during optimization and the ability to compare the resulting calculated spectrum, as a sum of the individual components, with the experimental spectrum. It is really easy to run the Line Fitting feature. Just click on the 'Auto Line Fitting' button of the ribbon. If the spectrum has already regions (i.e. integral regions or multiplet regions), it uses them to create the fit regions. If not, it creates the fit regions using the current "autodetect" configuration (under the integration options) to calculate the integral regions.
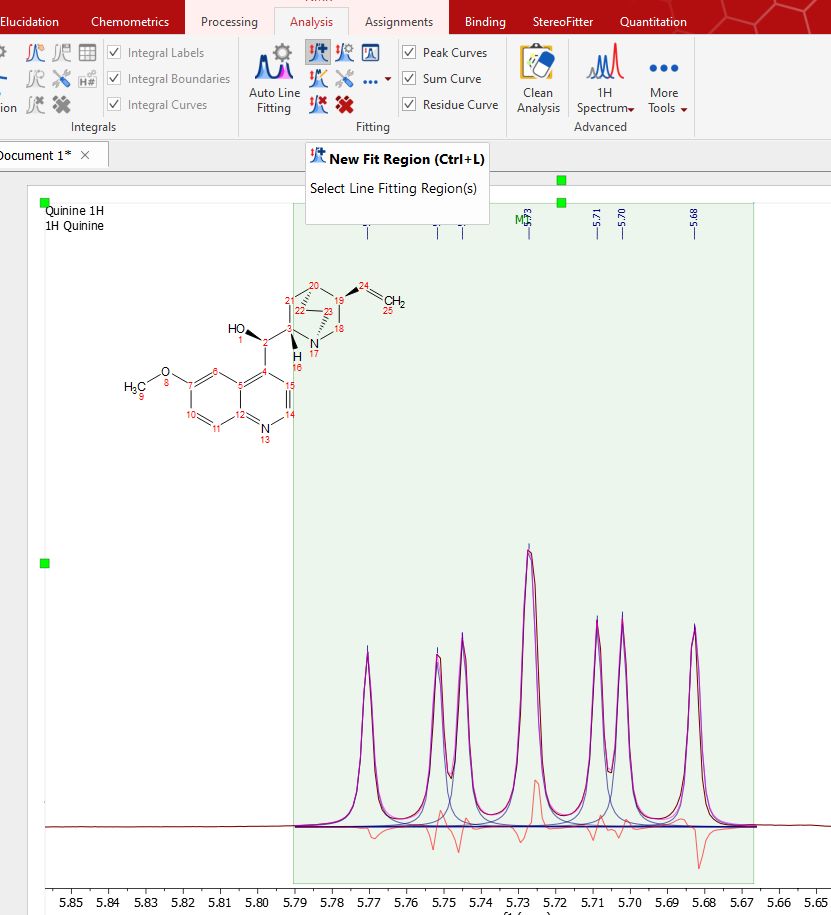
You can also add manual regions by clicking on the 'New Fit Region' icon NOTE: It you prefer, you could apply a previous peak picking before selecting the Line Fitting region. To have a better control over the peak picking, you could use the ‘Peak by Peak’ mode or the ‘Manual Threshold’.
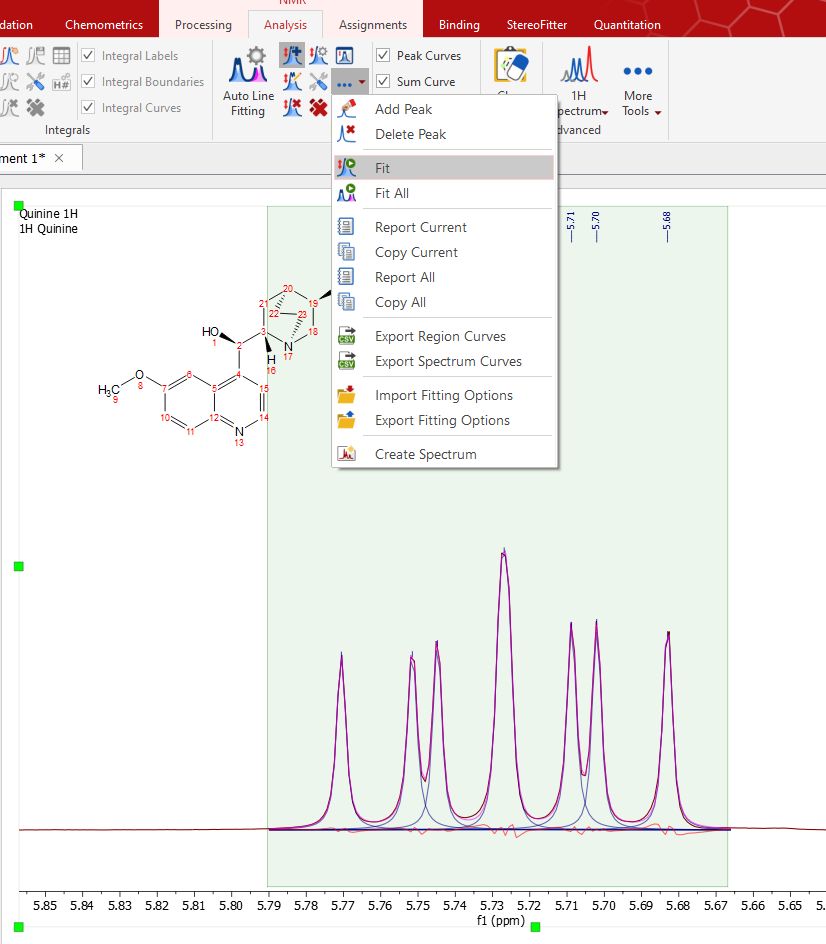
You can see in the picture above a blue line which corresponds with the fit peak, a red line which corresponds which the fitting residue and a magenta line which corresponds with the sum. Next, select Fit under the scroll down menu to carry out the Fitting:
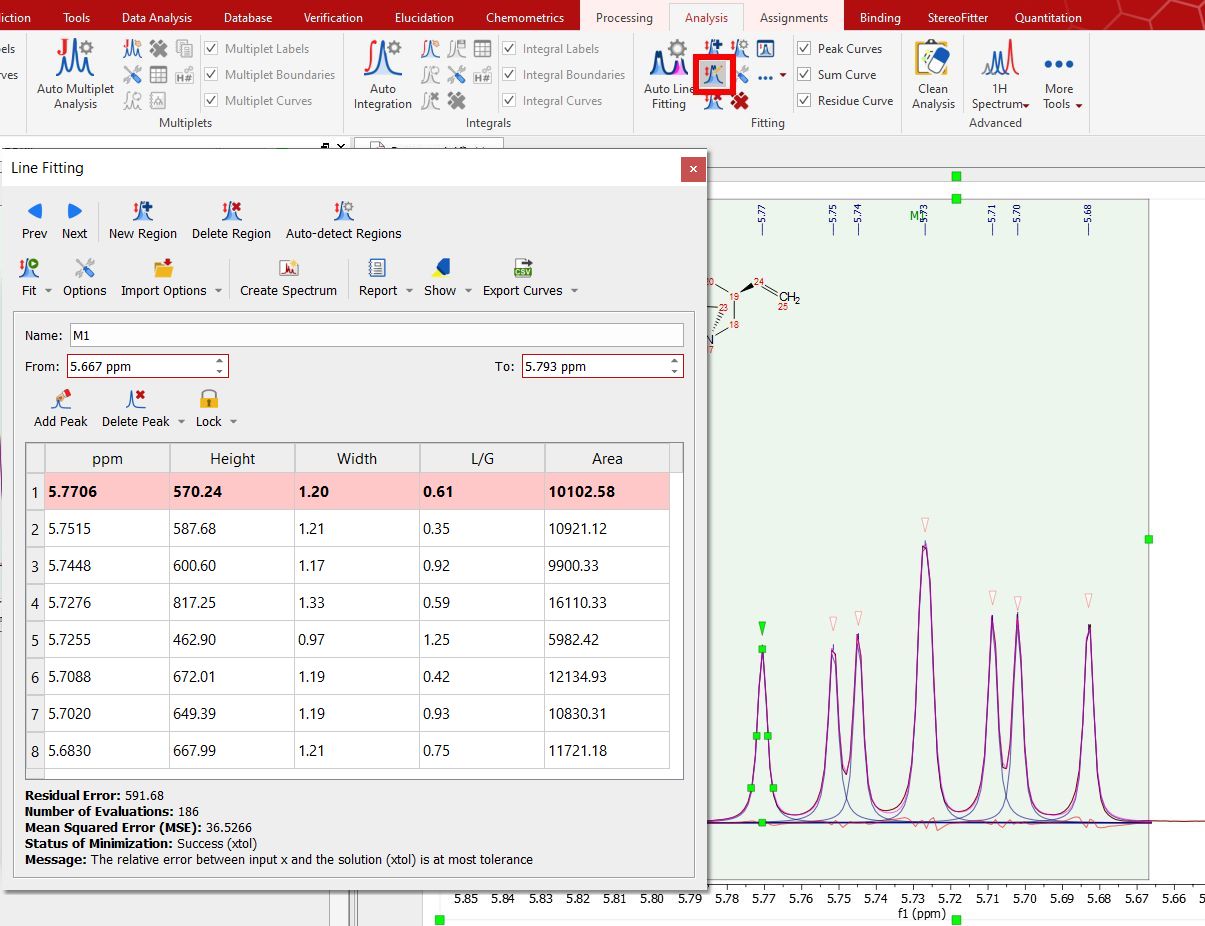
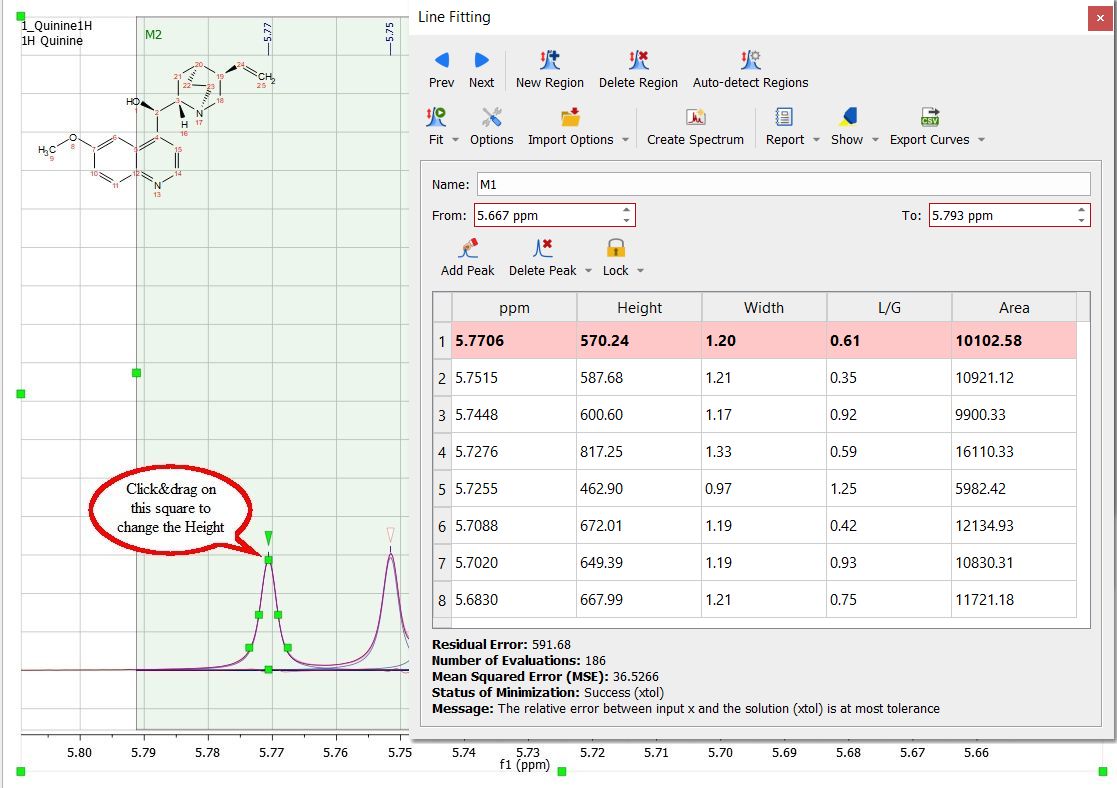
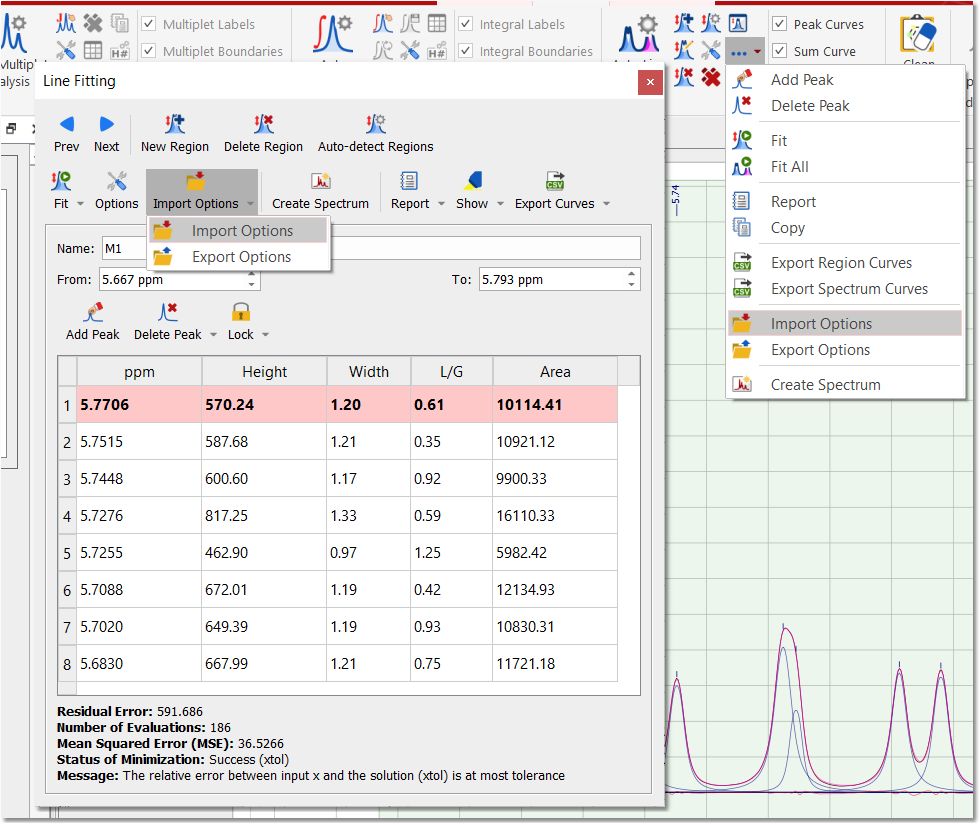
You will notice that the residue, the fit peak and the sum lines have changed. They constitute a starting point for the fitting (unless you are happy with the first fitting). If you are not happy with the initial fitting; you can add/remove components and change their parameters, just by selecting 'Edit Fit Region'. This will display the 'Line Fitting' panel, where you will find information about the peaks of interest (the chemical shift, in ppm; the Height, the Width, in Hz; the Lorentzian/Gaussian ratio and the Area).
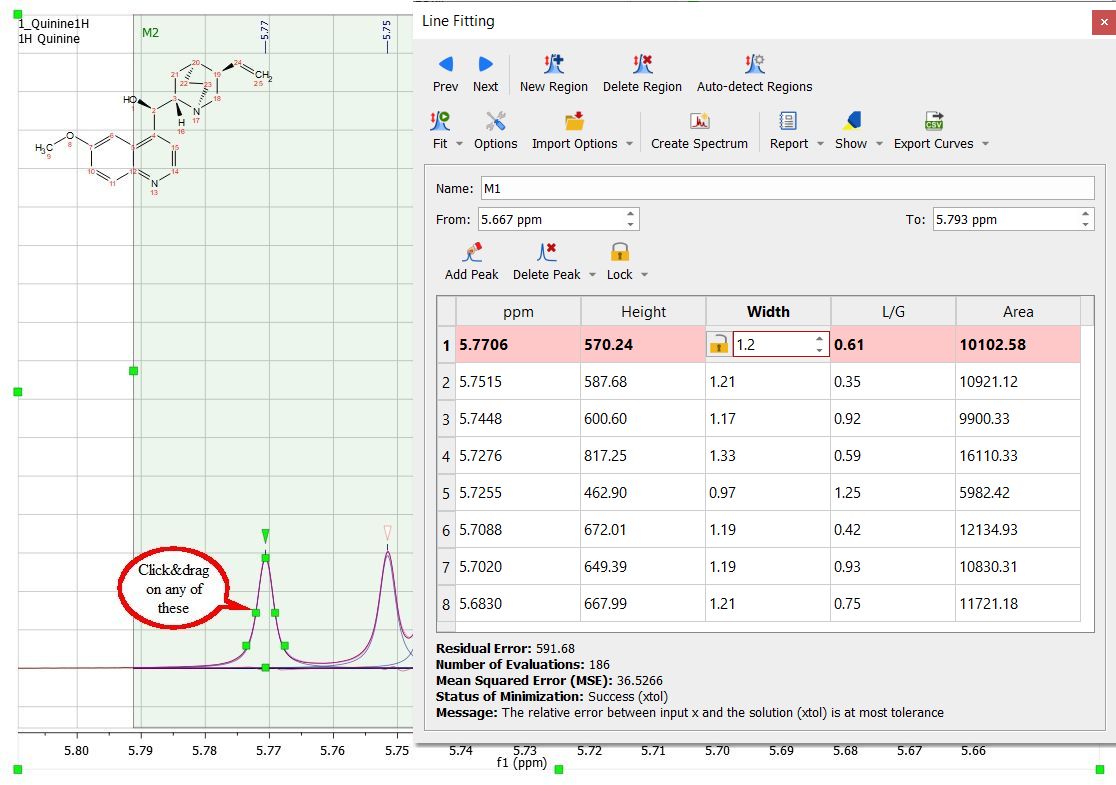
These parameters can be changed interactively over the spectrum (just by clicking and dragging over the green little squares) or by double clicking on each cell of the Line Fitting panel. Changes on any parameter will obviously affect to the 'Residual Error' (the smaller error, the better). For each point of the region, Mnova calculates the difference between the experimental and theoretical points, this value is powered by two and Mnova calculates the average of all the errors in the region of interest. Clicking and dragging the green square at the top of the peak, will change the Height of the peak.
The squares at the mid-height of the peak can be used to modify the Width of the peak:
Clicking and dragging on the next two squares will modify the Lorentzian/Gaussian ratio of the signal and finally the last square will be used to modify the chemical shift of the signal.
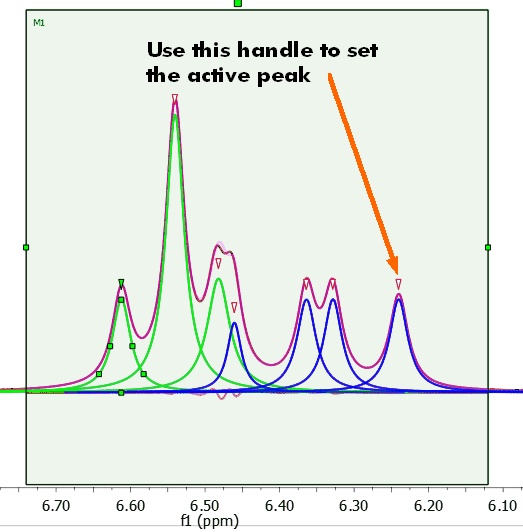
Clicking on the red triangles will set the active peak:
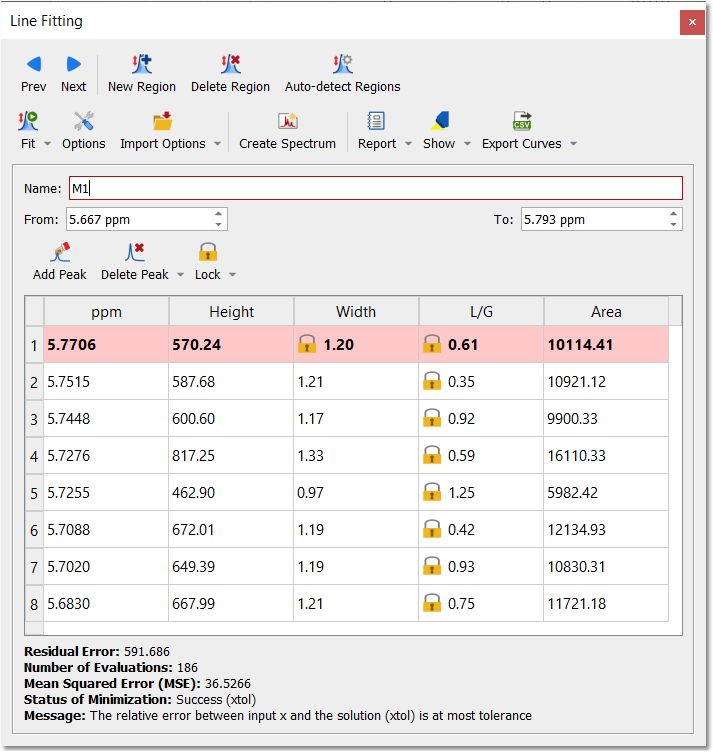
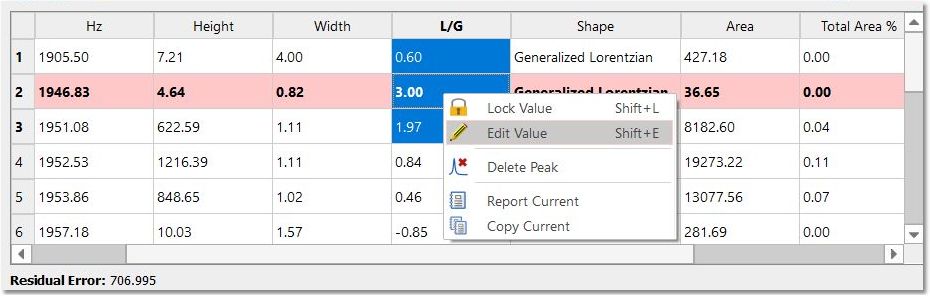
Any of these parameters can be locked for the Fit, just by double clicking on the applicable cell of the panel and closing the lock which appears. In the picture below it is shown how we have locked the width parameter of the peak 1 and also the L/G ratio of all the peaks, so these parameters will not change when the fitting will be applied. So a lock in front of a value indicates that it will not be optimized in the fitting.
You can also use the shortcuts below: SHIFT+L: locks all the selected values SHIFT+U: unlocks all the selected values SHIFT+E: opens a dialog to edit the selected values You can go through the table editing each cell by using the Enter key; which will display the editor dialog. Pressing the Enter key a second time, will change the value and close the editor. You can edit several cells together just by highlighting them, right clicking to select 'Edit Value' and typing:
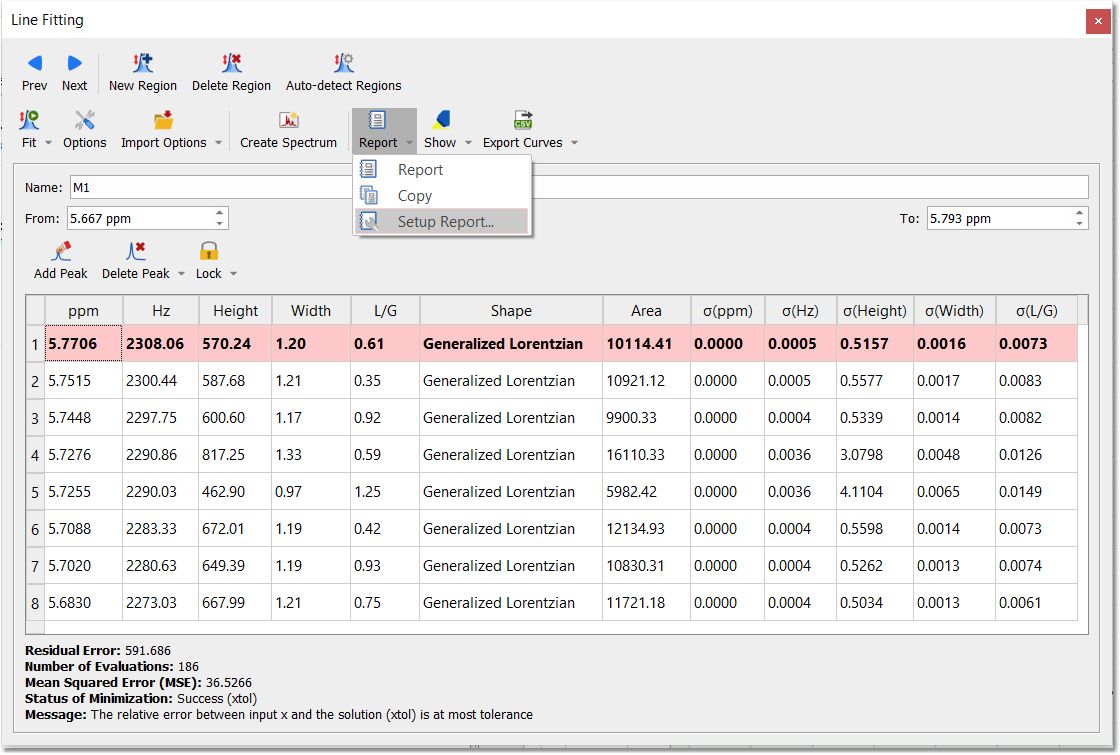
From the setup report dialog, you can display the standard deviation of ppm, Hz, Height, width, L/G and the peak shape:
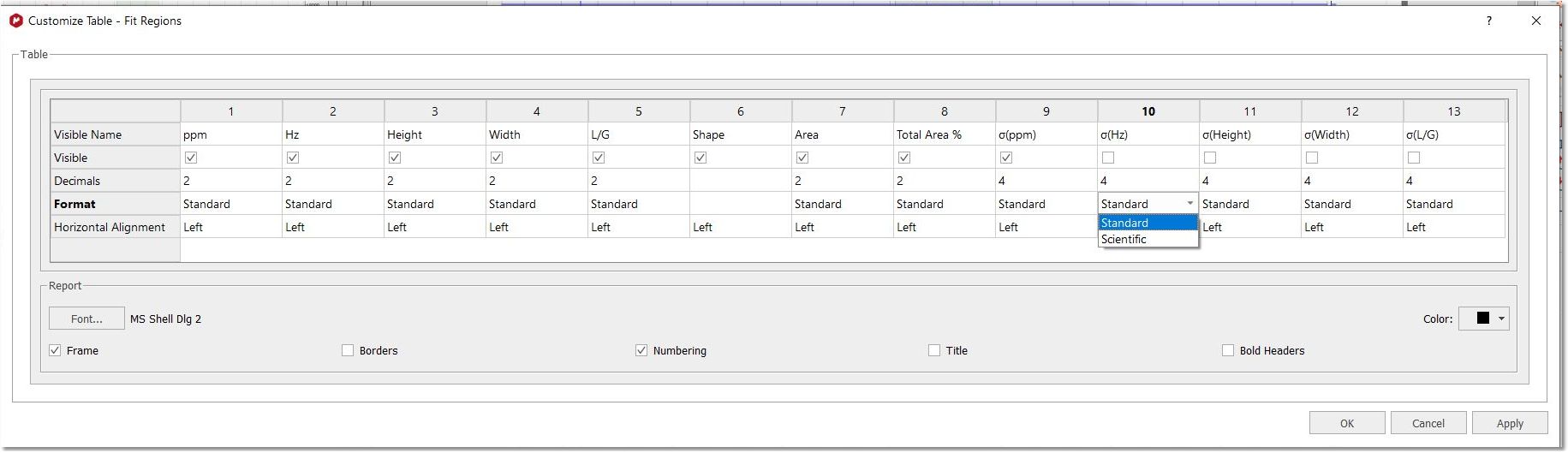
From the setup report dialog, you can also select the option to display the values in scientific notation:
Once you have run the fitting of several regions, you will find information about the residual error, number of evaluations, MSE (Mean Squared Error), status of minimization (the result of the fitting) and a message (explaining the meaning of the status of minimization).
L/G ratio: it does not pretend to be a mathematic representation. It is like a ratio between L and G. The value of the function to be fit would be: LG(x) = LGFactor*L(x) + (1-LGFactor)*G(x)
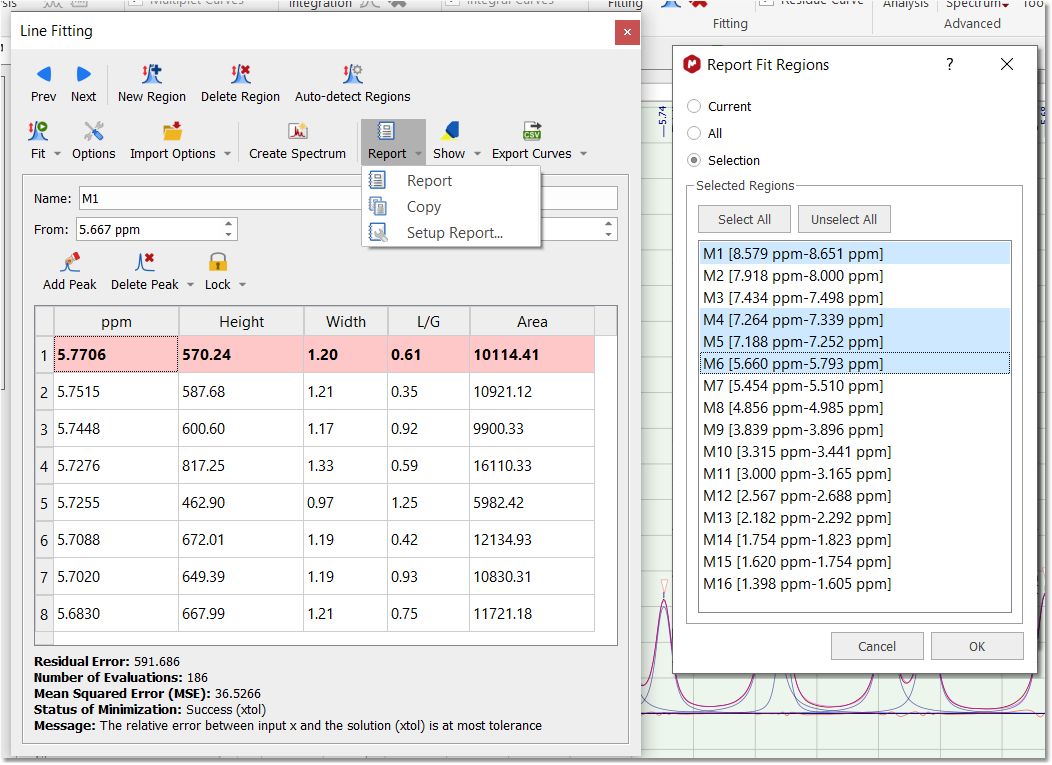
Of course you can fit several regions within a single spectrum, just by selecting a 'New Fit Region':
Once you are happy with all the parameters, you will only need to apply a new Fit, just by clicking on this icon
You can also use the Line Fitting feature for the simultaneous fitting across arrayed spectra. Just select the region and the fitting will will take place along all the arrayed spectra:
You can apply line fitting options and parameters for all stacked spectra.
Follow the menu 'Tools/Export/Fit Regions' to export the results in a .txt file.
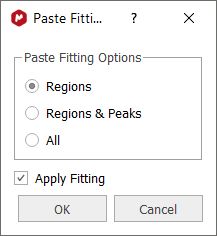
You can copy the Line Fitting regions from one spectra to another by selecting the first spectrum and doing 'Ctrl+C'; next jump to the second spectrum and follow the menu 'Home/Paste/NMR Fit Regions'. A new dialog box will be displayed with the options below:
- The Regions option pastes only the regions and fulfill them with an automatic peak picking. The regions are bounded to the current spectrum's range. - The Regions & Peaks option pastes the regions and their peaks, exactly as they are in the copied spectrum (same Shift, Height, Width, and L/G). - The All option pastes the regions, peaks and locks, exactly as they are in the copied spectrum. - The Apply Fitting option executes the fitting of the regions after pasting them.
Other Options of the Line Fitting panel The Line Fitting panel includes other tools, let´s see the meaning of them:
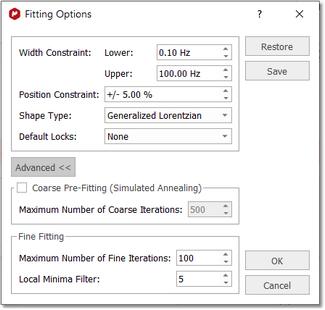
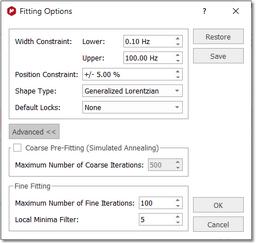
New Fit region: use this icon to create another region to be fit. Delete region: to delete the current region. Detect Line fitting regions automatically: it will apply an automatic Line Fitting over the regions detected by the automatic integration tool. Fitting Options: to display the Fitting Options window where the user will be able to establish some constraints for the fitting. Line Widths and Chemical Shift parameters can be constrained within user-defined values:
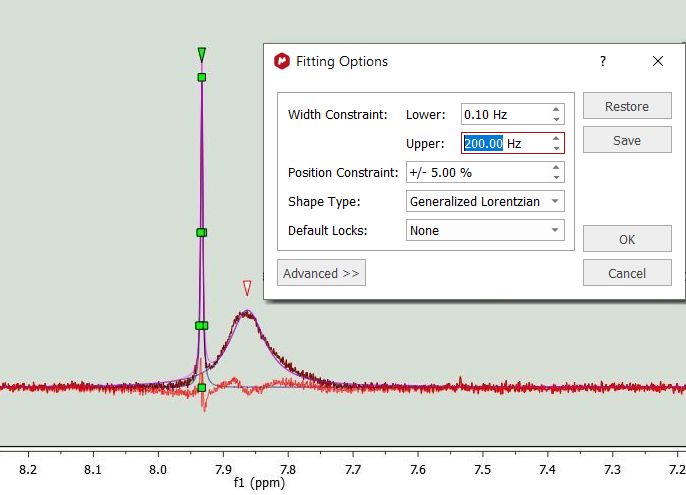
If the spectrum contains broad signals, just increase the Width Constraint to get good results. In the example below the Upper width constraint has been increased to 200 Hz:
Position Constraint: it takes into account the values outside the range (p=shift) p*(1-pos_constraint*p). A big value of the algorithm will 'shift' the peak far. Normally, you will not need to change the default value, because the peak should be close to the original position.
Shape Type: you can select between 'Generalized Lorentzian' or 'Lorentzian-Gaussian'.
'Generalized Lorentzian': The new generalized NMR peak shape profile proposed (which is a lorentzian plus a factor by another function), generalizes the ubiquitous Lorentzian shape. The mathematical expression (to be published) for the profile function contains only one extra parameter in addition to the three parameters of the Lorentzian line (location, height, and width). The new parameter does not interfere with any of the three 'classical' parameters (orthogonality) but affects the peak's kurtosis (for this reason we call it the kurtosis parameter). It is a-dimensional and has a 'natural' physical range of -1.0 to 2.0 (beyond these limits the formula is still operative but the peak profile starts splitting into a doublet or triplet). There are strong physical and conceptual reasons to expect that the new family of shape profiles will be much more useful in NMR spectroscopy than any of the often-used mixtures of Lorentzian and Gaussian shapes. In particular, we expect it to improve the quality of quantitative assays through NMR spectra and help the process of automatic editing of NMR peaks prior to structure verification.
NOTE: When a GSD peak picking is done the assumed peak shape is the Generalized Lorentzian.
By default, Mnova uses the so-called “Generalized Lorentzian” Lineshape which is similar but not equivalent to the linear combination of a Loretzian-Gaussian lineshapes. The type of lineshape can be selected in the Line Fitting options, as shown in the figure below:
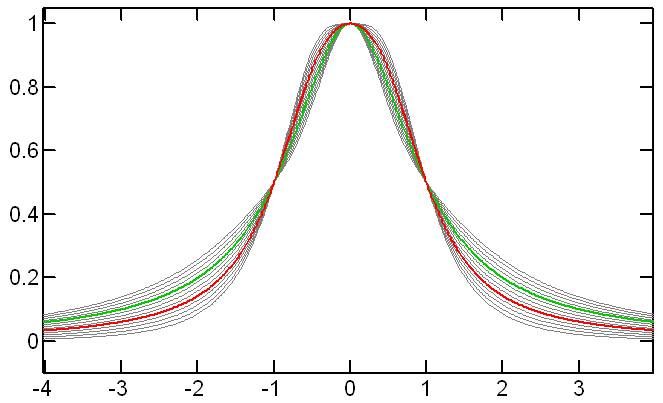
From a more theoretical point of view, to partially mitigate the unavoidable line shape imperfections found in NMR spectra, we have introduced the idea of a Generalized Lorentzian peak shape (GL). A single extra parameter describes the peaks shape deviation from the Lorentzian, which we call somewhat loosely the ‘kurtosis parameter’ (in the Line fitting dialog this corresponds to the L/G parameter which somewhat mimics the Lorentzian/Gaussian ratio, although this is not strictly the same, as discussed next). Briefly, the GL peak shape, though using just one ‘extra’ parameter, covers deviations from the Lorentzian about 3 times broader (in terms of kurtosis) and extending to both sides of the Lorentzian. This is illustrated in the figure below where the green line is pure Lorentzian, the red line is pure Gaussian (of the same half-height linewidth) and the gray lines are generalized Lorentzians:
When a linear combination of the Lorentzian-Gaussian lineshapes is used, the result is essentially an interpolation between the green and red lines so that, as can be seen in the figure, the actual range covered by this composite lineshape is more limited compared to the proposed Generalized Lorentzian. The GL thus provides more flexibility. At the same time because it is a rational function of the frequency offset, its imaginary part and its integral are easy to evaluate using exact, explicit formulas. This is not the case for a Gaussian line shape.
Mathematically, the Generalized Lorentzian (GL) function is defined by [1], where the amplitude has been assumed to be 1 and x corresponds to the frequency offset:
where k is the “kurtosis parameter”, so called because it affects the peak’s kurtosis and
As opposed to a L/G ratio which is within the [-1,1] range, the kurtosis parameter can take broader values, but they should usually be restricted to the [-2,+2] interval.
Default Locks: you can select from here what parameters you want to lock by default.
Coarse Pre-fitting (Simulated Annealing): coarse fitting algorithm which will look for random solutions, keeping the best one as starting point for the Fine Line Fitting in order to get a better solution. The user will need to select the Maximum Number or coarse iterations (to find the initial starting point). This algorithm will be faster but less precise than the Fine Line Fitting.
Fine Fitting: this algorithm uses a variation method to find the solution. With the initial parameters, it calculates the difference between the result and the real curve; after that, it modifies a little these parameters to achieve the real curve.
Local Minima Filter: The iterations stop when you reach the maximum number of iterations, when the difference with the real curve is too small, or when after several iterations, the variation of the parameters does not improve the fitting result. This parameter controls how much the parameters vary on each iteration. A high value means that the parameters change a lot on each iteration, which will be very useful in those cases where the initial curve is really different with the real curve. The user should not need to change this value. You can export (and import) the Line Fitting options from the 'Line Fitting ribbon' and from the 'Edit Line Fitting' dialog box:
Report: to report the Line Fitting panel. Selecting the 'Report' option will allow you to report the current region, all or a selection. From here you can also copy the report to the clipboard or customize the format of the report.
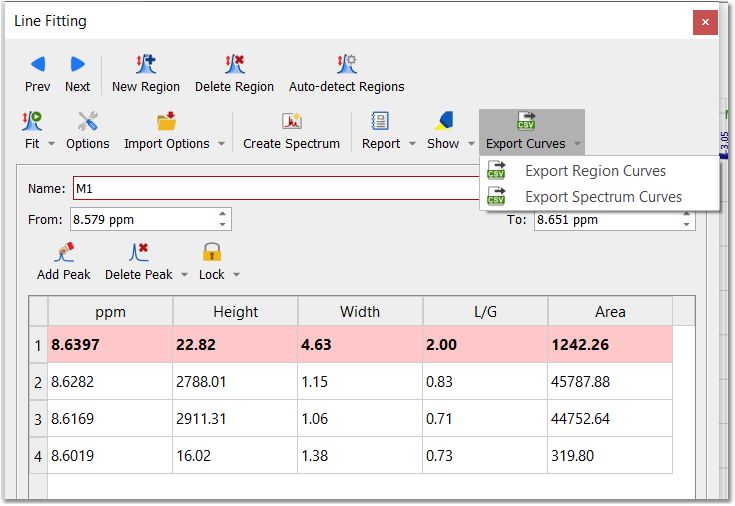
Export: to export the current fit regions or the entire spectrum curves as a CSV file.
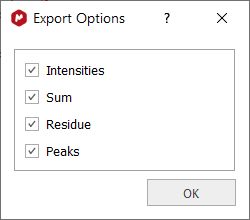
Once you have selected one of those options, you will have the capability to select if you want to include the intensities, sum, residue or peaks:
The resulting CSV file will have the column ppm under the "x" coordinates, and the respective columns with the "y" coordinates of those type of data selected (i.e. Intensities, Sum, Residue, Peaks). In the case of Peaks, there will be one column for each peak in the fit region / spectrum. The values are separated by commas.
Follow the menu 'Tools/Export/Fit Regions' to export the results in a .txt file.
Select Lines to Show: toggle button to show or hide the fitting lines (peaks, sums or residues) Previous/Next Region : to navigate through the fit regions. Name: to change the name of the fit region. From/To: to change the rang of the fit region. Add Peak: to add a new peak to the region Delete Peak: to delete the current peak of the region (or all peaks at the same time) Create spectrum: To generate a new spectrum from the fit regions. |