3D Molecule



3D Molecule |
|
|
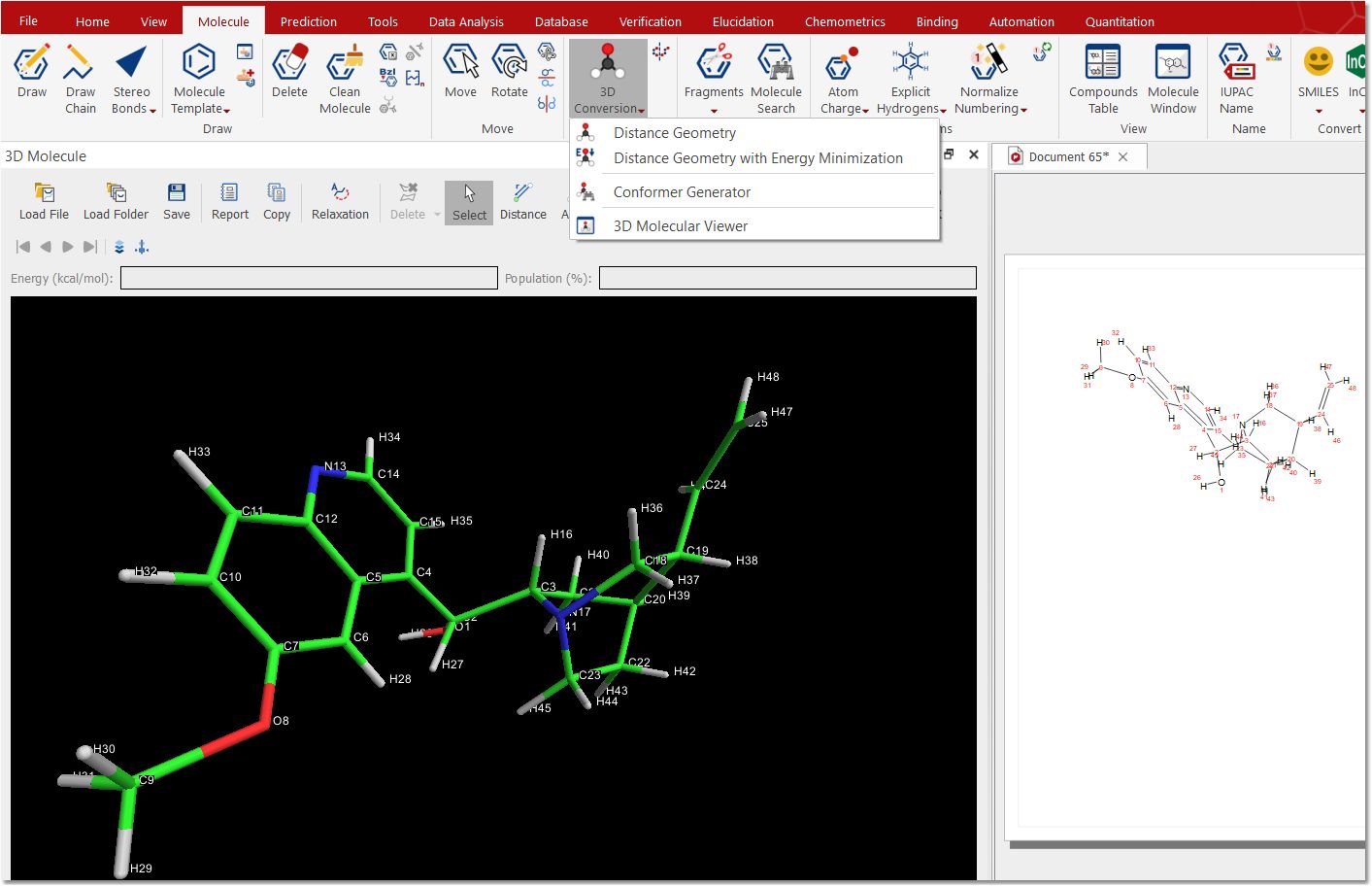
You can activate the 3D molecule viewer by following the menu 'View/Tables/Molecule/3D Molecule':
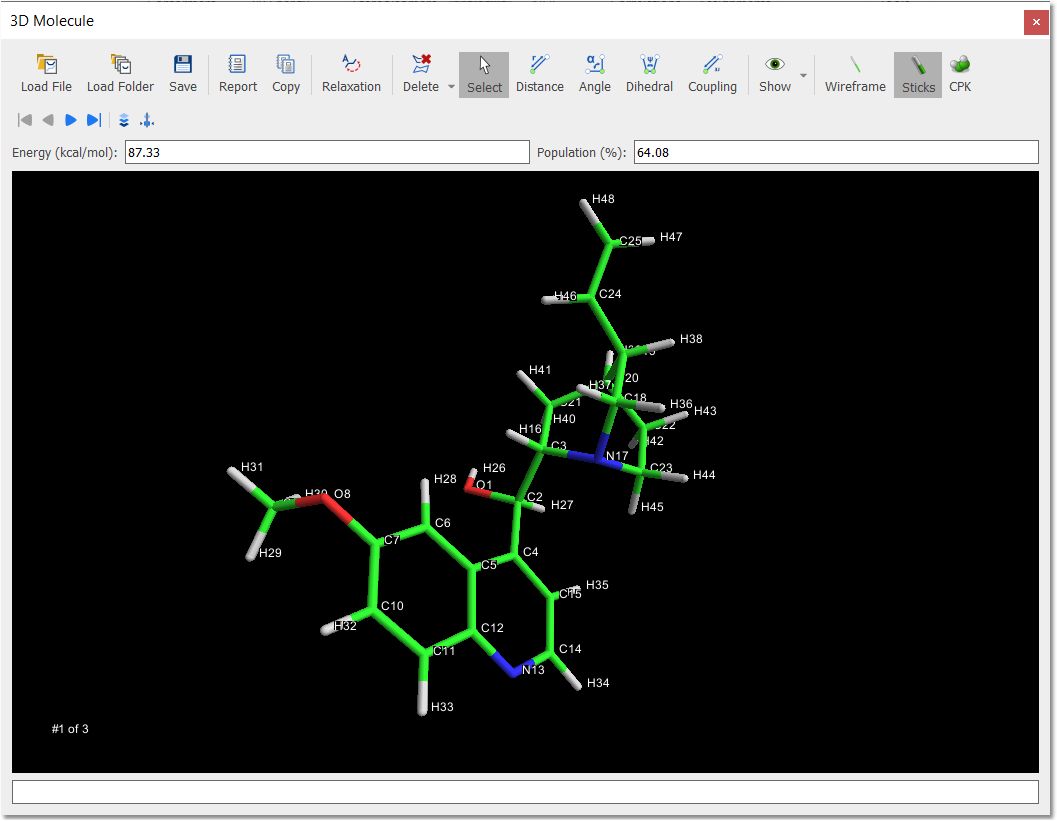
You will be able to navigate through the set of conformers by using the ‘Navigator toolbar’
Molecular 3D structures can be managed by:
-Clicking on the right mouse button and dragging (or scrolling up or down the mouse wheel) to zoom in/out the molecule structure.
-Clicking on the left mouse button and dragging to apply a TrackBall Rotation (the user is able to control the movement of a ball which contains the molecule).
From this panel, the user will also be able to select the rendering mode (Wireframe, Sticks and Ball Sticks).
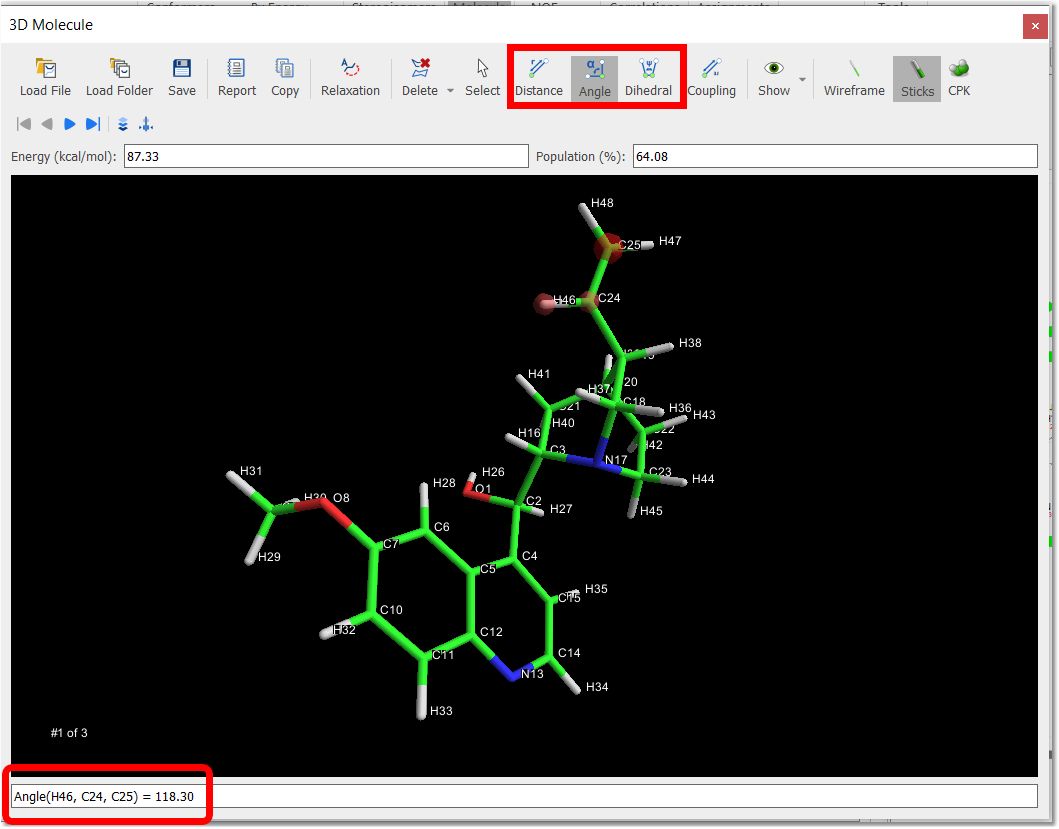
You will be able to measure bond distances, angles and dihedrals by just clicking on the corresponding buttons of the toolbar and next selecting the desired atoms to analyze.
Click on the 3D conversion feature button to convert a 2D molecule in a 3D structure, calculating the energy for each conformer and their population:
NOTE: If you are not getting the 3D molecule in the viewer, please change the OpenGL engine from the 'Preferences/Interface' dialog.
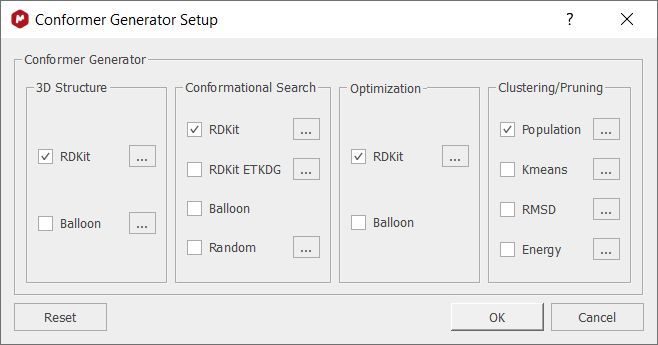
Clicking on the Conformer Generator option will allow you to select the algorithms for the calculation:
When loading a .mol file and using any of the available options, all possible conformers will be displayed in the 3D Molecule panel, including for each conformer: the Energy (kcal/mol) and Population (%).
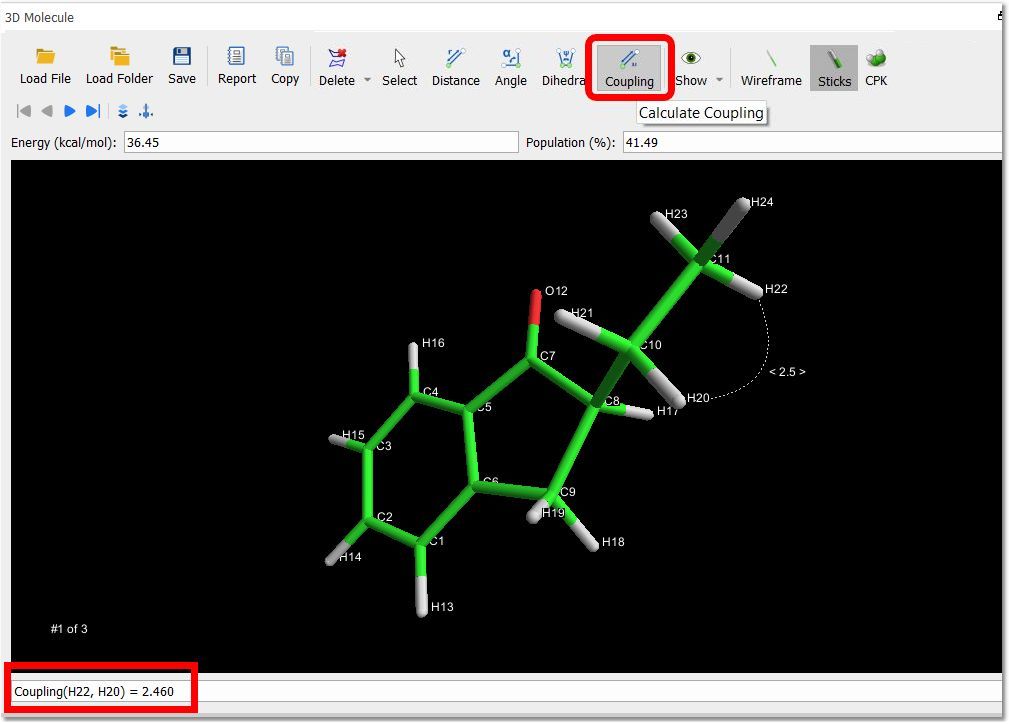
You can also calculate 3J[HCCH] couplings in sp3 carbons after having applied a 'distance geometry calculation'. Next, click on the 'Coupling' button, and select the two atoms of interest:
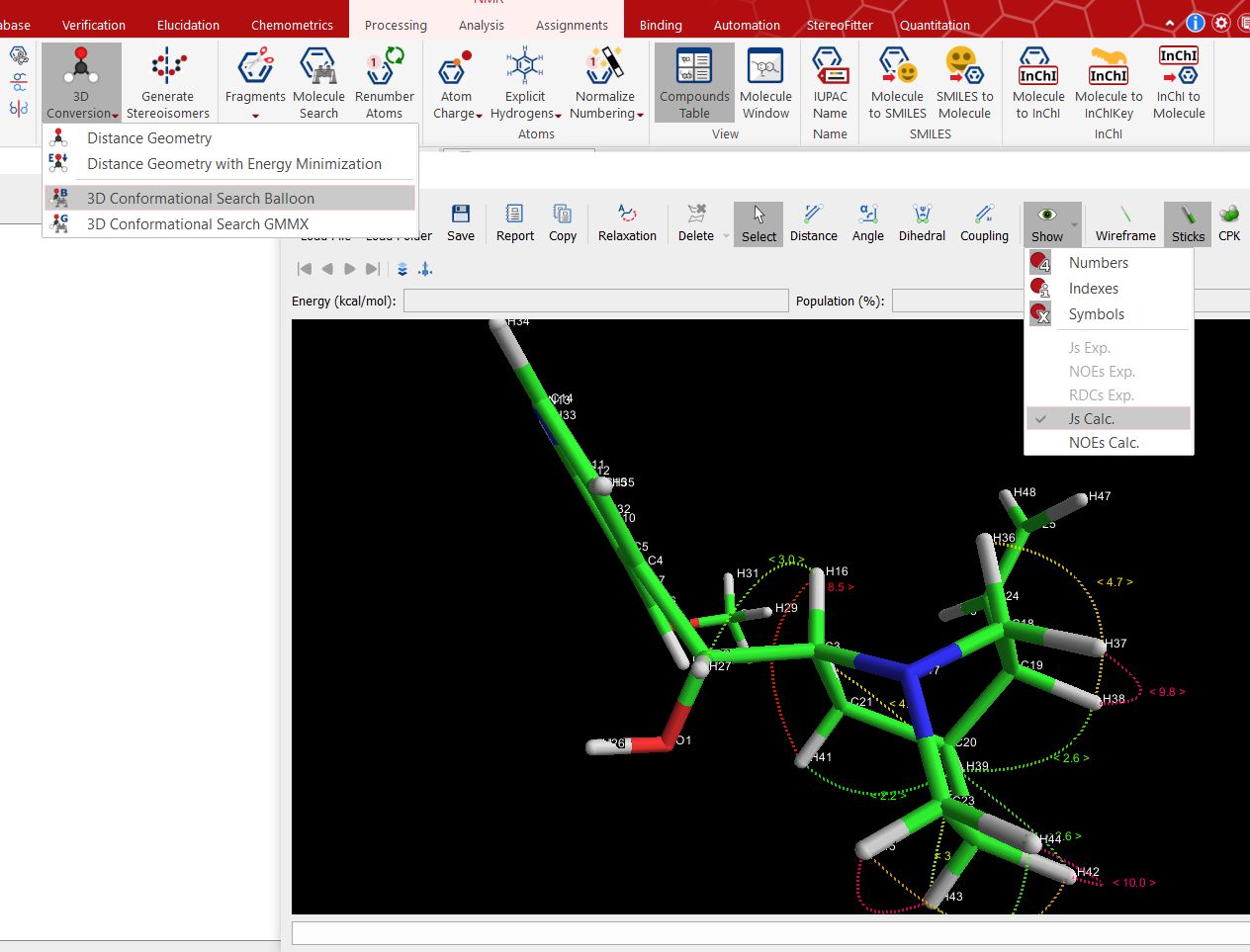
You can also calculate 3J[HCCH] couplings in sp3 carbons by doing a 3D conformational search with Balloon or GMMX analysis, making sure that you display the calculated J's values:
The J values and lines will be displayed with different colors (cyan to 0 Hz, green to 2 Hz, dark yellow to 4 Hz, orange to 6 Hz, red to10 Hz, magenta to 12 Hz, blue to 16 Hz and white to 20 Hz).
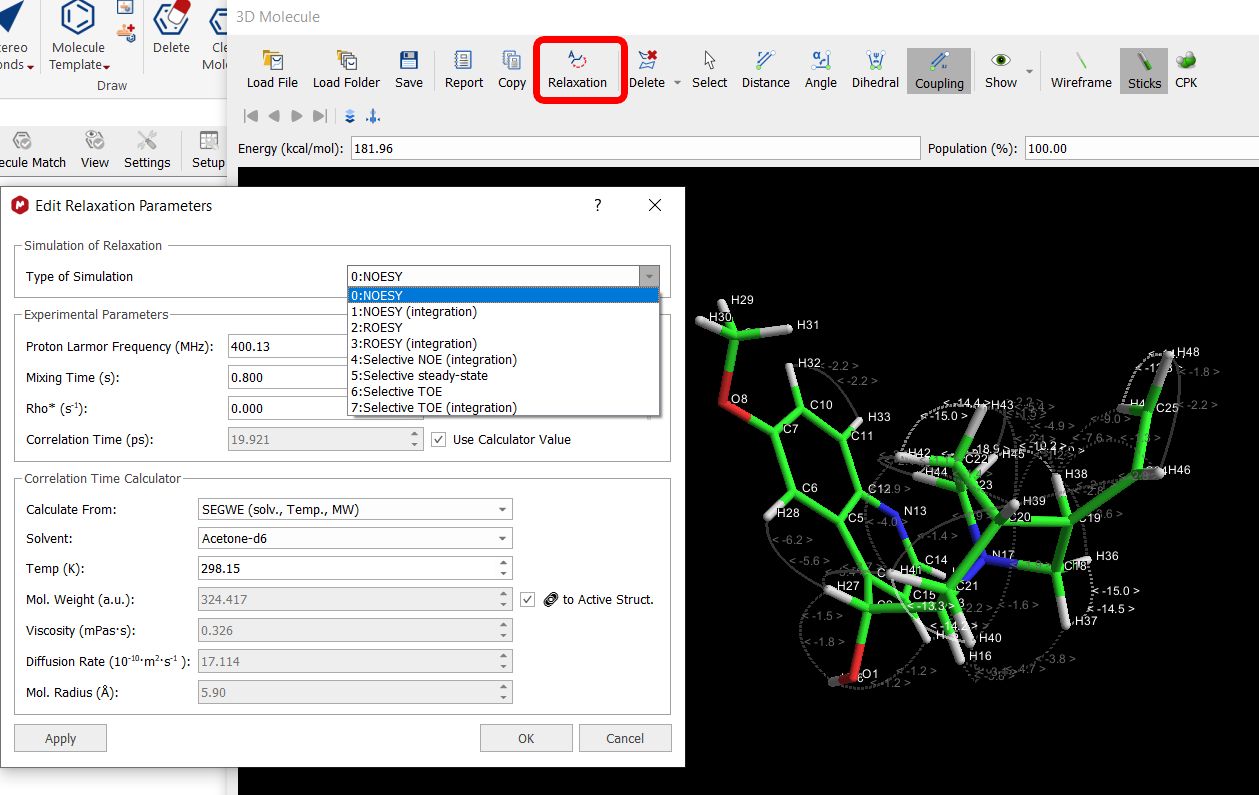
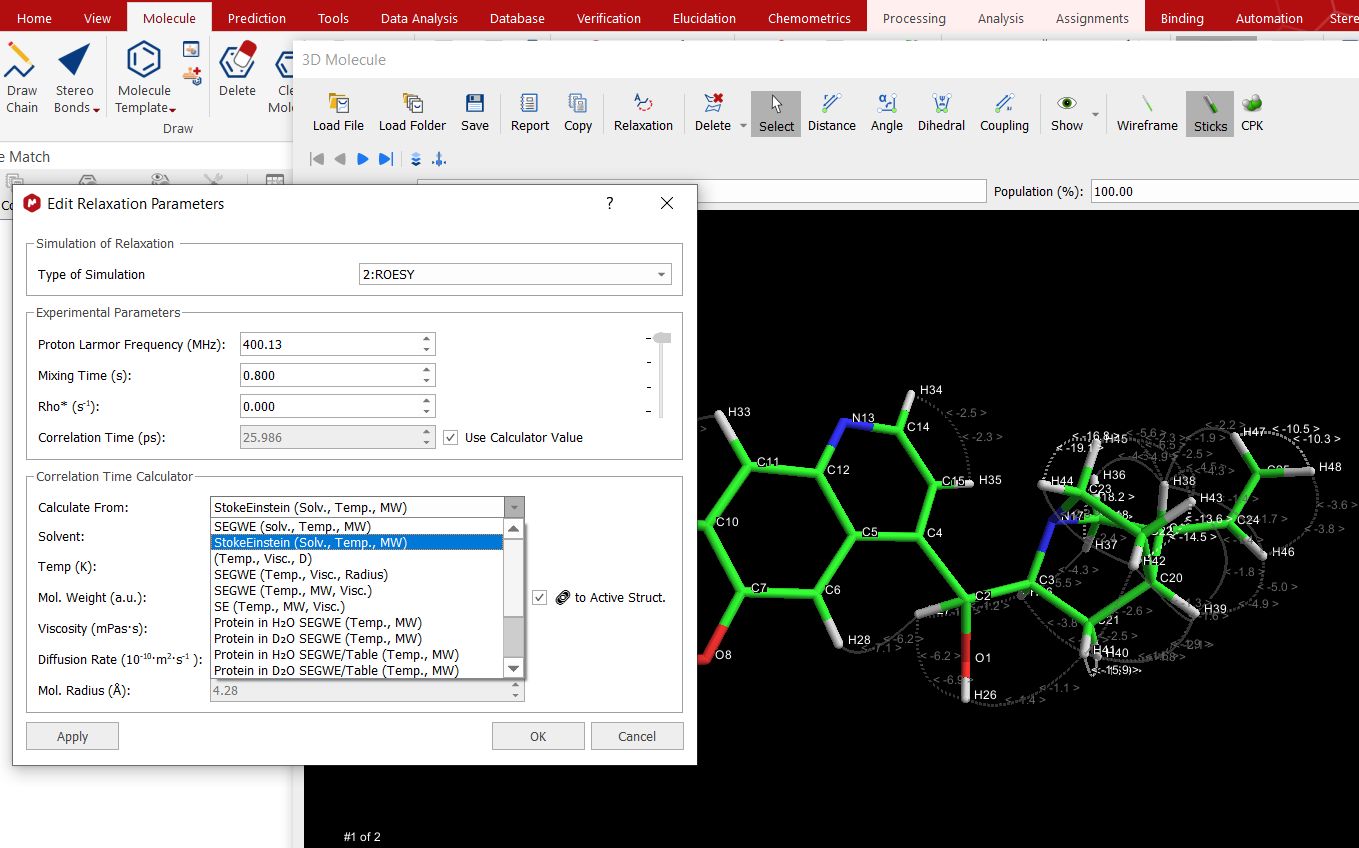
If your 3D molecule contains explicit protons, you can do relaxation analysis by clicking on the applicable button, which will display a new dialog to select the desired parameters:
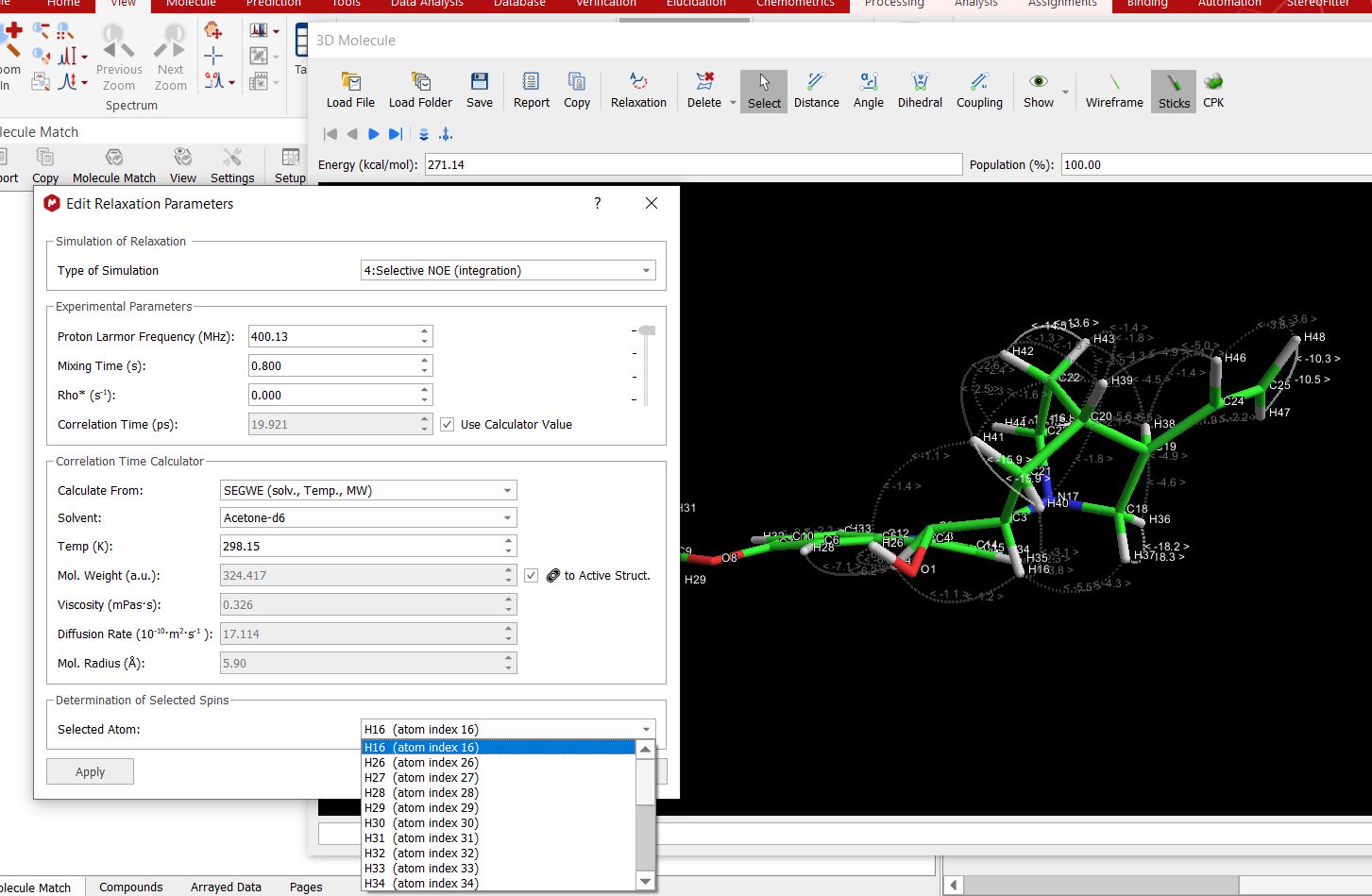
When choosing a “selective” simulation, an additional section named "Determination of selected spins" is shown at the bottom of the dialog:
The correlation time value can be added manually or it can be calculated by using the “Correlation Time calculator”, which is displayed when the option "use calculator value" is checked.
Once there you can select different methods, solvents, temperature, molecular weight, viscosity, Diffussion rate, molecular radius, etc...
When a method is selected in which the molecular weight is an input parameter, there is an option named "to Active Struct" next to the MW field. When this option is unchecked, the Molecular weight is editable.
The negative NOEs values will appear as white labels, the positive will be red and the absolute values less than 10 will be (grey) darker.
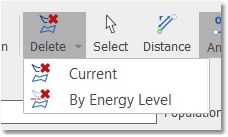
You could delete the undesired conformers just by clicking on 'Delete current conformer' button:
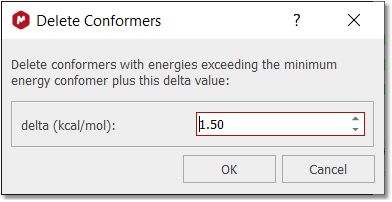
You can also delete conformers by energy level:
The '3D Conformation Search GMMX' tool will need a valid license for the Stereofitter plugin. |