How to carry out assignments?



How to carry out assignments? |
|
|
You will find the quinine datasets under the 'File/Help/Download Example datasets' menu)
You can carry out automatic assignments and atom to peak assignments both to 1H, 13C and HSQC spectra.
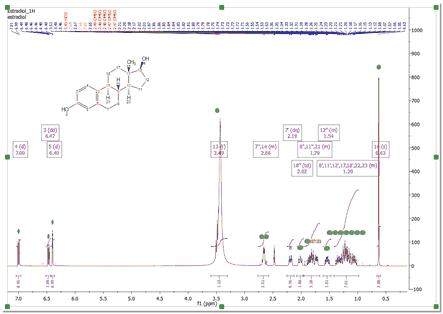
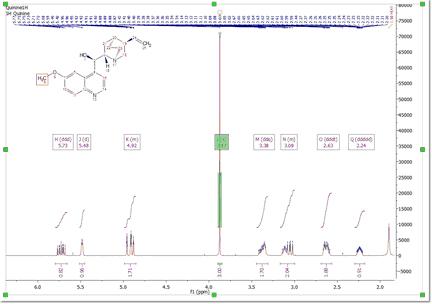
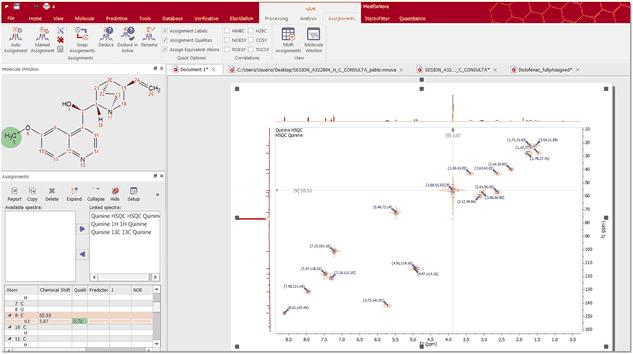
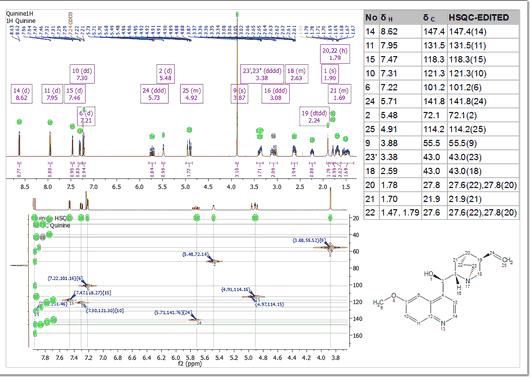
Automatic Assignment of NMR spectra has been a pipedream for many years, especially in the organic chemistry community. In spite of successful applications of computer algorithms for the automation of the processing and analysis of NMR spectra, fast and robust unsupervised assignment of NMR spectra of small molecules has so far been a discouraging endeavor. See this publication for further information. Mnova provides a very simple interface to automatically assign your molecule. Just load your NMR spectrum (1H, 13C and HSQC; alone or combined) with the applicable molecular structure and follow the menu 'Assignments/Auto Assignment':
Mnova will automatically run a multiplet analysis and will fully assign your spectrum with the applicable molecular structure. If you have a license of the prediction plugin, the assignments will be more accurate.
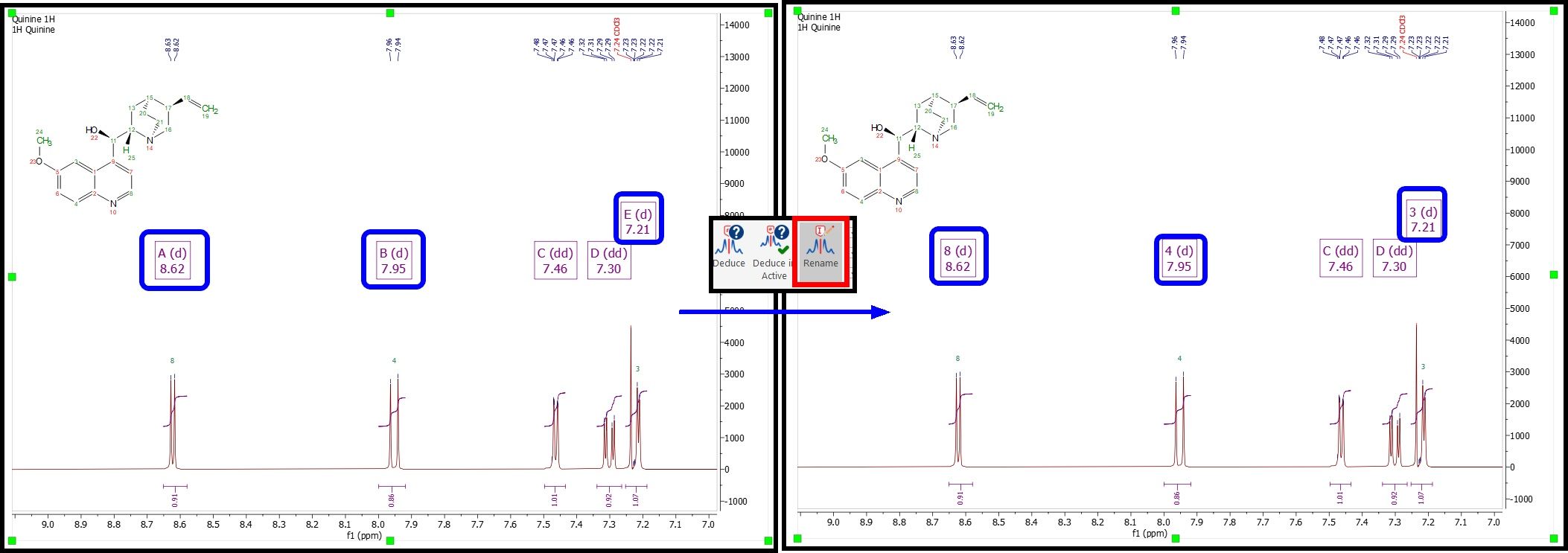
The multiplet boxes will be renamed with the corresponding atom number of the molecule and you will get an assignment label with the atom number over the applicable peak (including a colour quality coding). In addition, hovering the mouse over the atom will highlight the applicable signal in the spectrum:
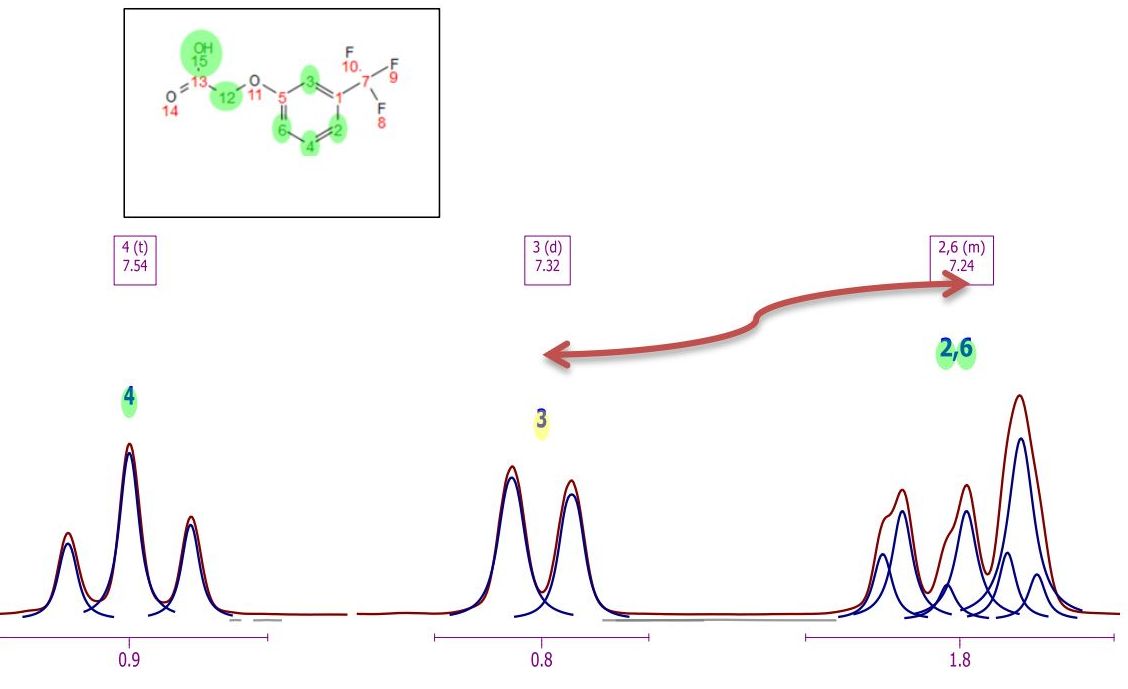
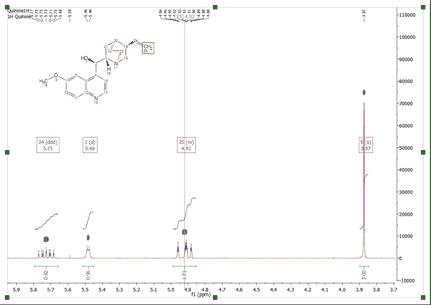
This algorithm for automatic assignment of NMR spectra has to be understood as a quick method to generate an initial ‘rough’ assignment that in some cases will require manual intervention from the user to easily amend any potential incorrect auto-assignment. The auto-assignments could assign, for example, all the protons properly except two which are swapped (as illustrated in the figure below) where assignments (3) and (6) must be swapped.
You can swap assignments by following the menu 'Analysis/Assignments/Swap Assignments' (shortcut: S). Then you will only need to click on one assignment label (or multiplet box) and drag and drop it to the target assignment label (or multiplet box).
If the target multiplet has more than one assignment, you will be asked for which assignment must be used for swapping.
The assigned chemical shift can be also modified by moving the assignment labels. In 2D spectra, it is possible to move vertical and horizontal assignments. When moving assignments to a position inside a multiplet range in the 1H spectrum the new assignment will be linked to the multiplet. The new assignment will be the multiplet shift or the selected position depending on the option "Use always 1H multiplet shift when adding 2D assignments".
MANUAL ASSIGNMENTS Mnova provides a very simple interface to manually assign your molecule. Open your NMR spectra in the same document and load/draw the molecule structure. Next, follow the menu 'Assignments/Manual Assignments' (or use the shortcut 'A').
Finally, click on an atom in the molecular structure (or a spectrum region) and then release the mouse and drag it to your desired peak, peak range, multiplet box or integral curve. We recommend you to assign your atoms to your multiplet boxes in order to transfer assignments through datasets.
The assigned chemical shift is added (automatically) to the assignments table when assigning to a multiplet, integral, peak or position, either in 1D and 2D spectra.
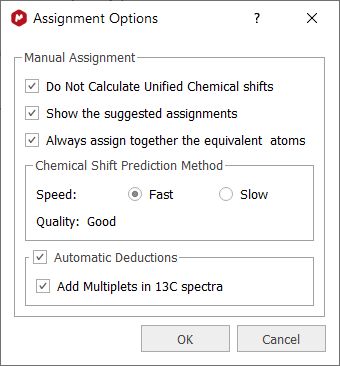
With the 'Assisted Assignment mode' (which works for 1H and 13C spectra having a multiplet analysis), a 1H and 13C predictions are made in the background. When hovering the mouse over the atoms of the molecule, the suggested multiplets in the spectrum will be highlighted (following a color code: green, yellow, red, gray- depending on the quality: grey: -1 to -0.5; red: -0.5 to -0.25; yellow: -0.25 to 0.25; green: 0.25 to 1) and viceversa if you hover the mouse over the multiplet boxes, the suggested atoms in molecule and compounds table will be also highlighted. This feature can be disabled from the 'Assignments options'.
This multiplet will now be assigned to the atom (which will turn to green). Once the assignment has been made, you will get an atom number label on the chemical shift (with quality colour coding). Hovering the mouse over the atom will highlight the applicable peak in the spectrum and hovering the mouse over the peak will highlight the corresponding atom on the molecular structure. To customize the assignments labels, click here.
If you do not want to show the assignments labels or the quality colour coding; just uncheck the applicable option from the 'Assignments Properties' dialog box. From the Properties, you will also be able to customize format of the correlations labels.
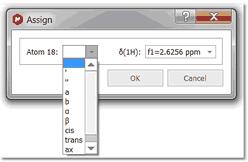
The user can also assign a region of the spectrum just by clicking, dragging and releasing the mouse over the desired region. In the example below, we have assigned a -CH2 group. If you want to assign the diastereotopic protons, you would need to hold down the 'Alt' key when assigning, so a new window will be displayed to allow us to select which atom we want to assign, 18, 18', both (in blank) or even we can select any other annotation: 18a, 18b, cis/trans, ax/eq, α/β :
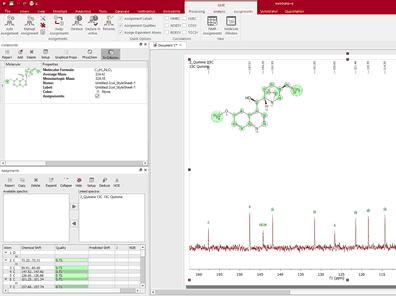
You can assign a 13C-NMR spectrum of the same molecule in the same document. To do that, select the atoms from the 'Molecule Window' panel by following the menu 'View/Panels/Molecule Window' (or from the Compounds Table; from 'View/Tables')
To keep the assignments propagation, follow the menu 'View/Tables/Assignments' and make sure that the desired are linked (in the example below, we have selected the 13C).
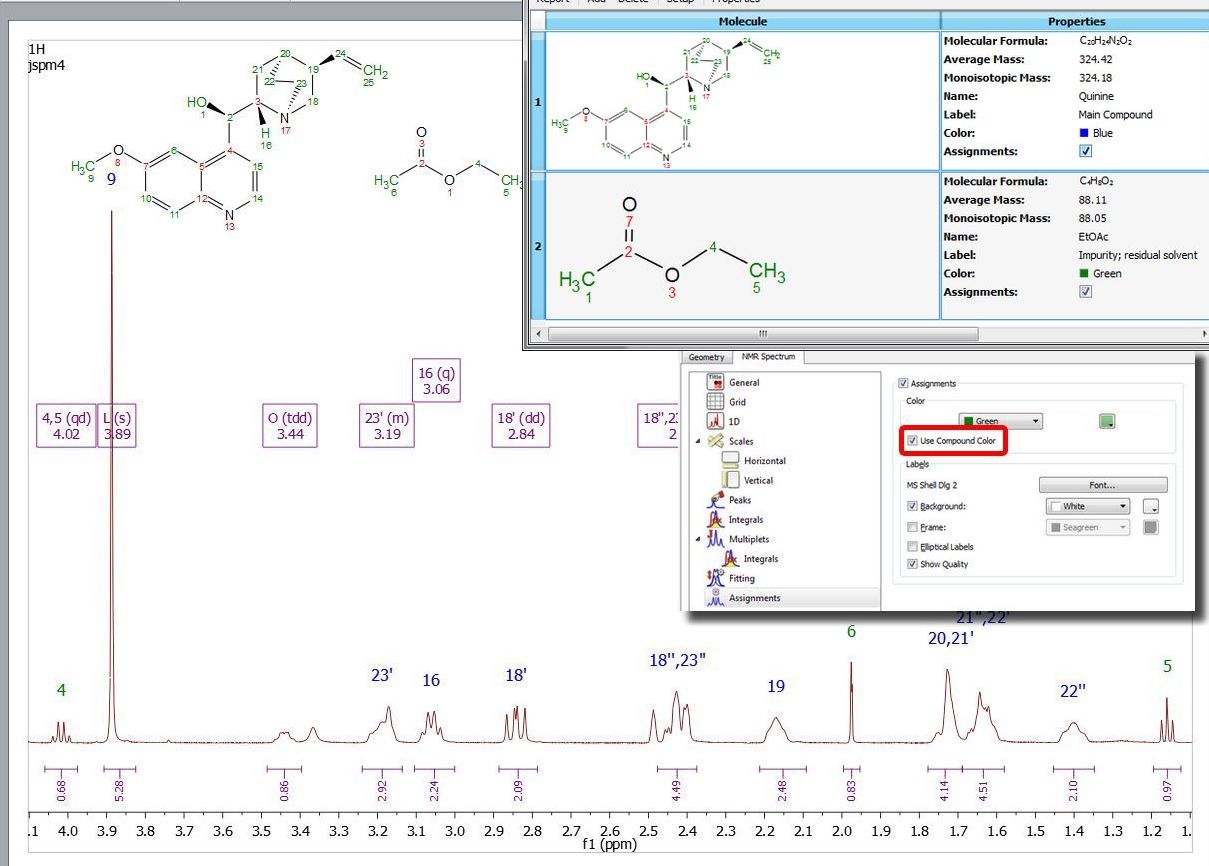
If your document has more than one molecule (see also this tutorial), use the compounds table to select a different colour for the assignments of each compound. In the example below the assignments labels of the quinine appears in blue and the assignments labels of the EtOAc are green (make sure that 'Use Compound color' is checked in the Assignment Properties):
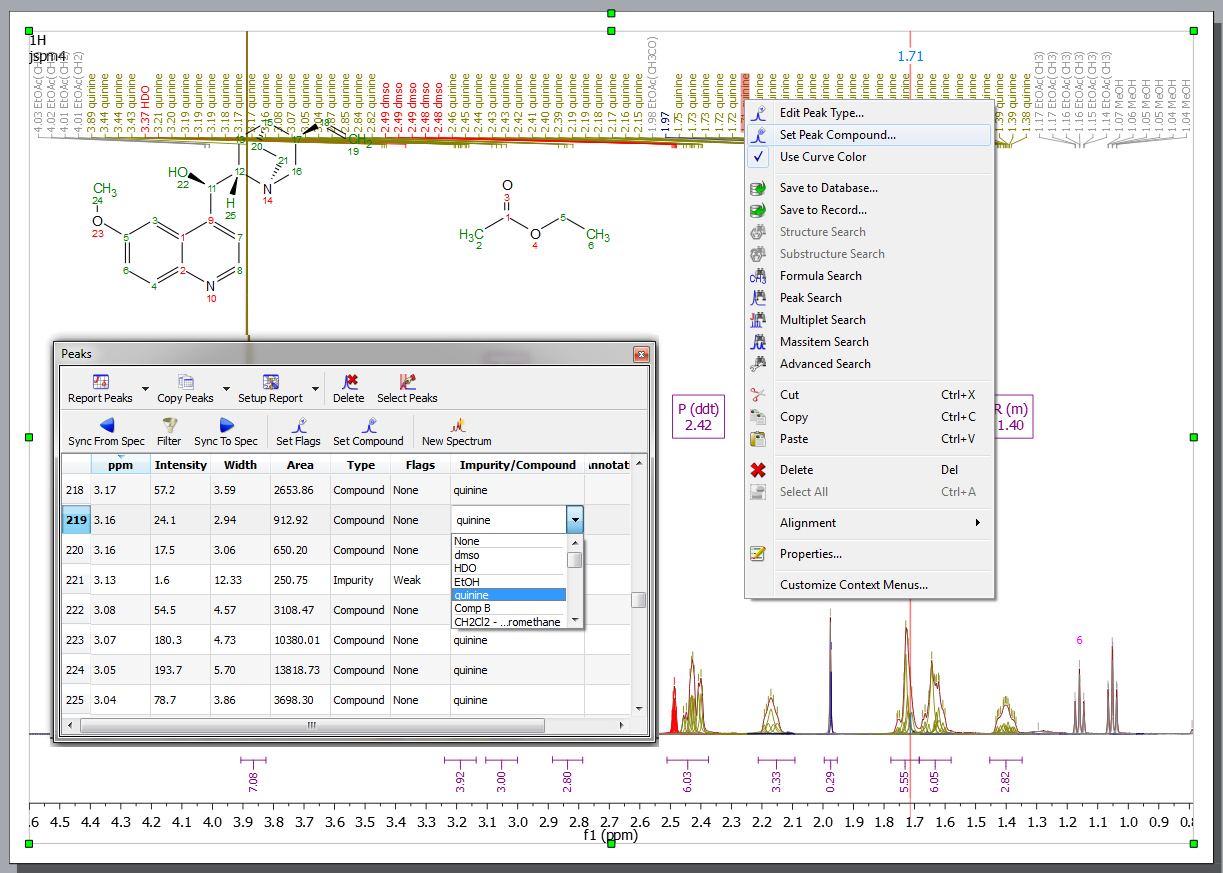
If your document contains more than one compound, you can select the compound for each peak from the Peaks Table (by clicking on the 'Set Compound' button or by double clicking on the 'Impurity/Compound' cell). The same effect will be obtained by right clicking on the Peak label and selecting ‘Set Peak Compound’ (please make sure that the 'Use Curve Color' box is checked in the 'Peaks Properties'):
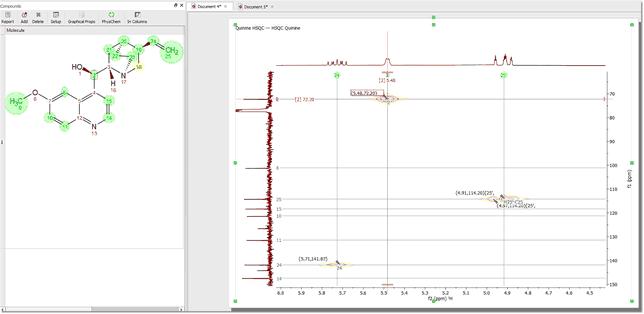
Assignments in 2D Once you have assigned the 1H and the 13C spectra, if you jump to a 2D-NMR spectrum (in the same Mnova document) you will see the assignments graphically on the screen.
From the Assignments options, you can use the unified chemical shifts when you assign a 2D spectrum. With the option "Do not calculate unified chemical shifts" is off, the unified shifts are calculated taking into account all the shifts provided by the linked spectrum objects (multiplets/peaks/integrals), either when the assignments are made manually, by moving an assignment and getting a new correlation deduced or from other deduction routines. When you assign to a position (in a 1D or 2D spectrum), all multiplets in the document corresponding to this/these chemical shift/s will be assigned (the assignment for the selected atom will be the shift and range of the multiplet with the narrowest range). Note: the highlighting would show all the linked objects because all of them are considered for the unified shift calculation.
When the option "Do not calculate unified shifts" is on, each assignment to a spectrum object adds a link, so several links are still allowed. But no unified assignment is calculated. The assignment is taken from the last mutliplet/peak/integral assigned. Note: only one linked object would be highlighted, which is the one used for the assignment value of each atom.
Basically when the: a) Do not Calculate Unified Chemical Shifts is checked: it doesn't matter if you assign first to the 1H or HSQC, both assignments will be taken into account, and the resultant chemical shift will be the one corresponding to the narrowest assignment range of the two assignments. b) Unified Shifts is unchecked: the assignment can change when you assign to 1H or HSQC, it will be always the last assignment done.
From the dialog above, you can also select the prediction algorithm used for the 'assisted assignment mode' and you will have an option to always assign together the symmetrical atoms (this option will also appear as a check box in the Assignments ribbon)
When the 'Automatic Deductions' option is not checked:
oassignments are not deducted in 2D/1D spectra when the user assigns manually a 1D/2D spectrum oassignments are not deducted when an assignment label is moved.
When the Deduce button of Assignments Table is pressed, the automatic deduction routines are executed, independently on this option is checked or not.
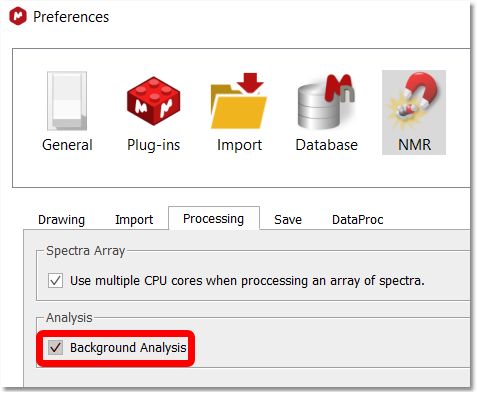
The "Add Multiplets in 13C spectra" tool needs the "Background Analysis" enabled (in Preferences/NMR/Processing) to work. This is a general feature, valid for all 2D spectra, not only for HSQC spectra, when the user assigns to a peak or multiplet.The assignments made in the 2D peaks will be transferred to the existing multiplets in the 13C spectrum. If there are no multiplets in the 13C spectrum, they will be deducted, created and assigned automatically using the background information. (this behavior is produced only when the option Background Analysis is checked, and the new option "Add multiplets in 13C spectra" is also checked).
The searching of the 13C-multiplets compatible with the 2D assignment will be done in a different way according with the Use Global Assignment option: - Global Assignment ON: the 13C-multiplet is auto-assigned (or created and auto-assigned in case of background multiplets) only if it's the unique 13C multiplet with a shift inside the range 2D chemical shift ± 0.25 ppm. -Global Assignment OFF: only the 13C-multiplets whose range contain the 2D chemical shift are automatically assigned (or created and autoassigned if background multiplets are needed).
To carry out assignments to 2D spectra, type the applicable number of the atom in the 'Assignments Table' or graphically select the atom in the molecule structure and the applicable signal (peak label, chemical shift or multiplet box) in the 2D:
Once an assignment is made in a 2D spectrum, this assignment will be transferred to the applicable multiplet box in the 1H spectrum.
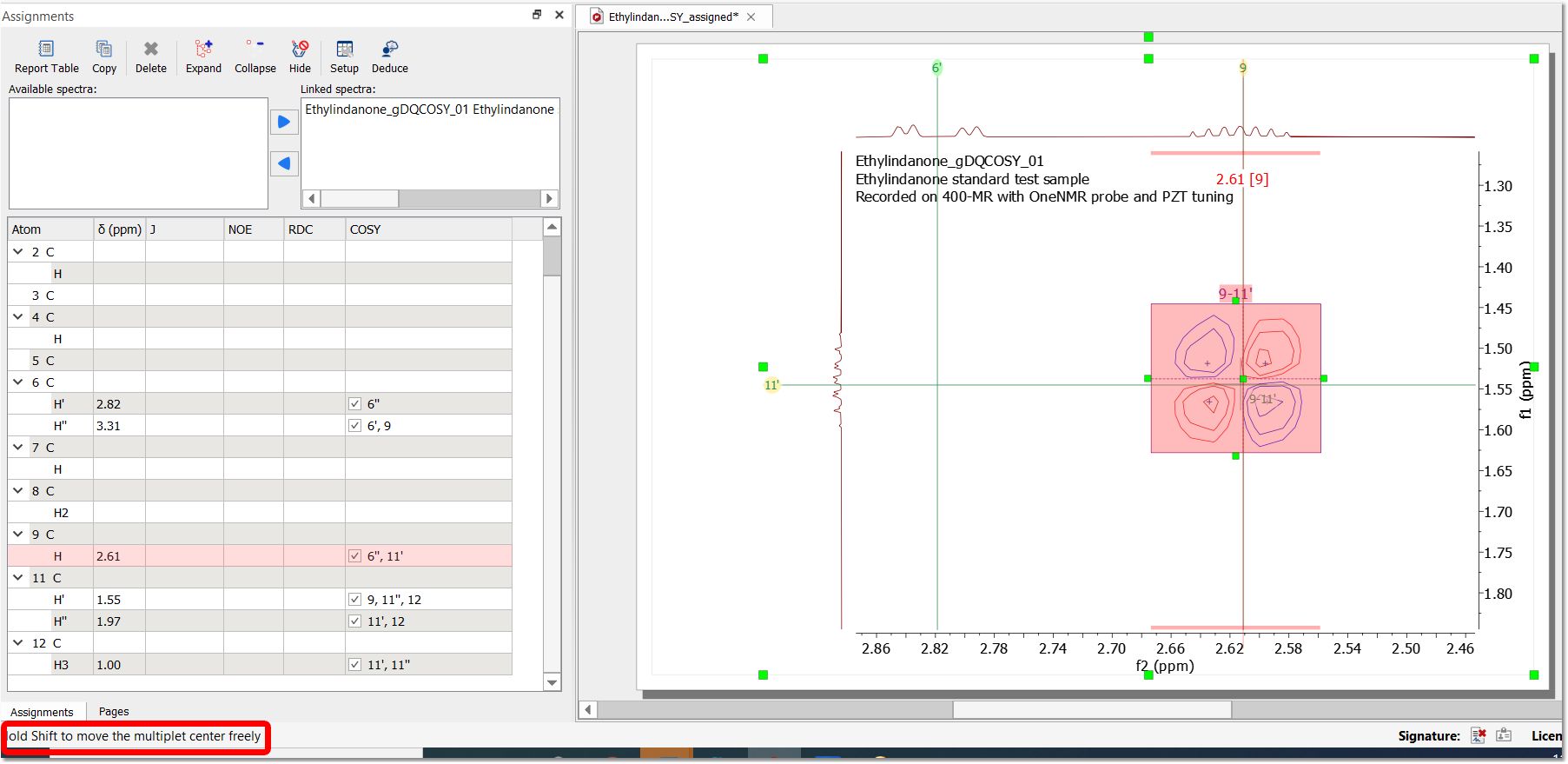
When hovering the mouse over the 2D multiplet area, a handler at the "center" of the multiplet box will be displayed to allow you to modify graphically the f2 and f1 multiplet shifts of the multiplet. When moving it towards a gridline, it snaps to the gridline; by holding down the Shift key, it will allow you to to freely move the position (a message is displayed in the status bar):
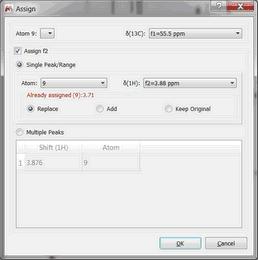
If you had already assigned the atoms in the 1D datasets and you assign in the 2D, the assignment will be replaced. You can force the visualization of the Assign Dialog by pressing the Alt key together with the second click of the mouse (when assigning); so you will be allowed to keep the assignments from the 1D, replace or add new assignments:
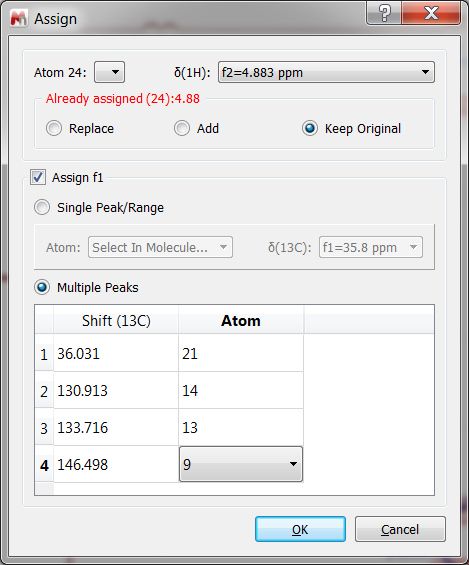
If you are working with a HMBC, you could assign one of the protons and then all the 13C in the same step (by selecting 'Multiple Peaks' from the panel):
Alternatively, you could assign only one of the carbons and the next times that you need to assign carbons to the same proton, you will not need to select again the applicable proton (as it is already assigned). If you have overlapping in the proton dimension, you could hold down the Alt key and click to get the 'Assign' dialog displayed which will allow you to select any other proton.
When you manual assign to a 2D Multiplet: •Assignment shift and range are set to the multiplet shift and range. oWhen the option 'Always assign together the equivalent atoms' is checked, the assignment shift is made to the 1H multiplet shift (when it exists). •The 2D multiplet name is updated in order to contain the assigned atoms. oThis name is not updated if only one dimension is assigned. •A link between the assignment and multiplet is created: oWhen the assignment is deleted (one dimension), the atom name is removed from the multiplet name. oThe multiplet name is updated when the corresponding correlation is removed, graphically from the spectrum or from Assignments Table. oWhen an assignment label is moved (horizontal and vertical dimension) and the final position corresponds to a 2D multiplet, the assignment get linked to that multiplet. ▪When the option 'Always assign together the equivalent atoms' is checked, the assignment shift will be the corresponding 1H multiplet shift (when it exists). Assignment Range will be the 2D multiplet range in all cases. oWhen 2D assignments are swapped, involved multiplets are updated. The user has the option of swap also (or not) the HSQC correlations. ▪Swapping can be done using the assignment labels (horizontal and vertical) or using the 2D multiplet boxes. oWhen the assignment shift is modified from the Assignments Table: ▪if the new shift belongs to the multiplet range: The assignment range is not modified. The user will be asked if these changes should be applied also to the multiplet: •YES: the multiplet shift is changed. The link is maintained. •NO: The multiplet is not modified. The link is broken, so correlations, constants, etc. are removed. The assignment is made to the new shift. ▪if the new shift is outside the multiplet: the link is broken, the assignment range is calculated using the default margin value (0.01). (The multiplet shift and range are not modified. The multiplet name is updated.) oWhen the multiplet is removed, the assignment is deleted (both dimensions).
RENAME When this button is pressed, the active spectrum multiplets are renamed with the assigned atoms information (a new link will be added between each multiplet and the corresponding nucleus). That button will be enabled only when the active molecule is assigned and there are multiplets in the active NMR spectrum (and both, molecule and spectrum are linked).
It can be used for 1D and 2D: In 1D: the multiplet will be renamed only if the assignment chemical shift is inside the multiplet range. In 2D: the multiplet will be renamed only when each shift of the two dimensions are inside the corresponding multiplet range, and the correlation is already defined.
Existing assigned multiplets remain unaltered. Chemical shifts, ranges and qualities could change, as a new link is being added to the link list.
If "do not calculate unified chemical shift' option is OFF: the chemical shift, range and quality will recalculated taking into account the new link (the assignment will correspond to the narrowest assigned linked object). Background prediction will be executed if there is no prediction data (needed for the assignment quality calculation).
If 'do not calculate unified chemical shift' option is ON: the assignment would have to be taken from the last multiplet/peak/integral assigned.
As it's assumed that the user wants to maintain the current assignments as they are, the chemical shift, range and quality will not be modified.
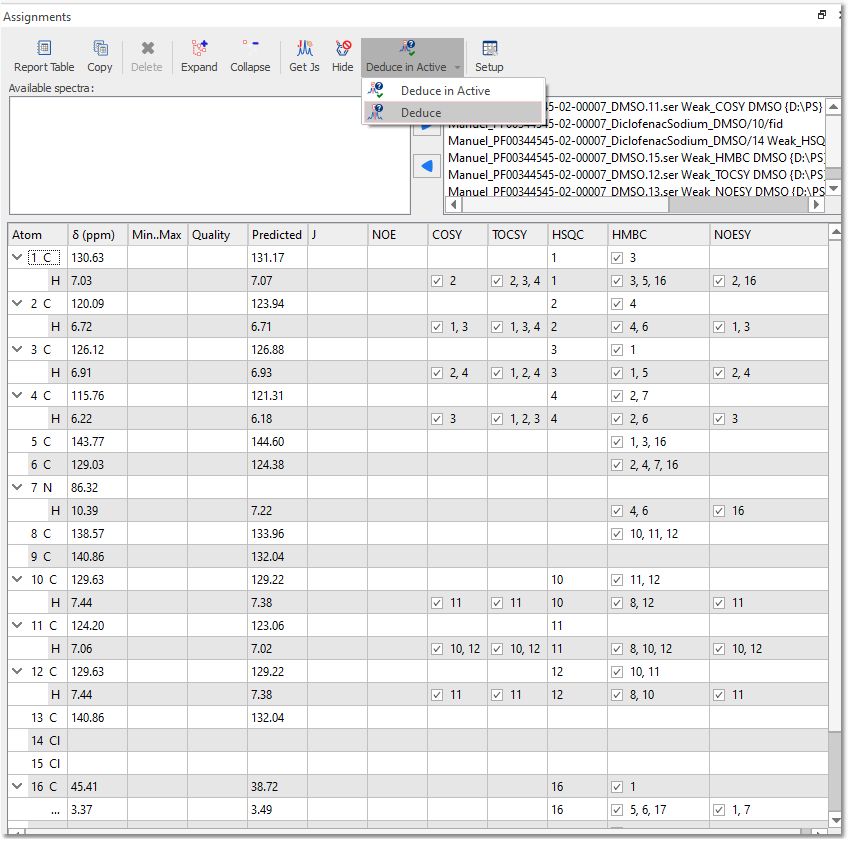
DEDUCE TOOL Mnova provides this tool to deduce 2D correlations by following the 'Assignments/Deduce' Menu. You will need to firstly assign the 1H and the 13C NMR spectra (with multiplet analysis in the 1H) and apply a Peak Picking in your 2D plots. Finally, click on the 'Deduce' button to get the correlations in the 'Assignment table' for each 2D experiment (COSY, HSQC, HMBC, NOESY, etc..):
'Deduce in active' is only available when there are assignments in the active molecule.
So the assignments behavior is different depending on the assignments that you have in F1 and F2: - If you do not have any assignment; the first assignment that you will make will go to F2 and the next for F1. - If the peak is already assigned in one of the dimensions, when you make the assignment, Mnova waits only for the selection of one atom (in the dimension which is not assigned) in order to complete the information. If you do not want to do it, you would need to do ‘ALT+click’ to launch the ‘Assign dialog’. - If the peak is already assigned in both dimensions and you try to assign it to another atom; the ‘Assign dialog’ will appear (as Mnova does not know what you need to do).
There is a 'Background Analysis' check box under the 'File/Preferences/NMR/processing' menu which could be useful for your assignments.
If it is checked; during the Manual Assignment mode, the Autodeduction routines create (and assign) on the fly the background multiplets when the 'real' multiplets are not defined by the user:
oWhen assigning to any 2D spectra (to multiplet/peak/range), the corresponding 1H multiplets are automatically created and assigned using the ranges of the background multiplets. o2D multiplets of any spectra are automatically generated and assigned when assigning to a 1H multiplet. oIf a Carbon was autoassigned in the previous point, this carbon shift is used to generate and assign the 2D corresponding multiplets in the other spectra. oThese routines analyze first the multiplets defined by the user, then, the background multiplets, and then, if these last are not found, the peaks defined by the user. •Background multiplets are also analyzed in all 2D spectra when pressing the action Deduce Assignments. In this case, as the background analysis just starts when pressing the button, the program waits always until this analysis has finished in all spectra. (Or until the waiting time limit of 5 seconds has been reached). •The background multiplets are also analyzed in all 2D spectra when the user moves an 1H assignment label to a 1H multiplet. The program waits until the analysis has finished (in all spectra) or the 5 seconds has been reached.
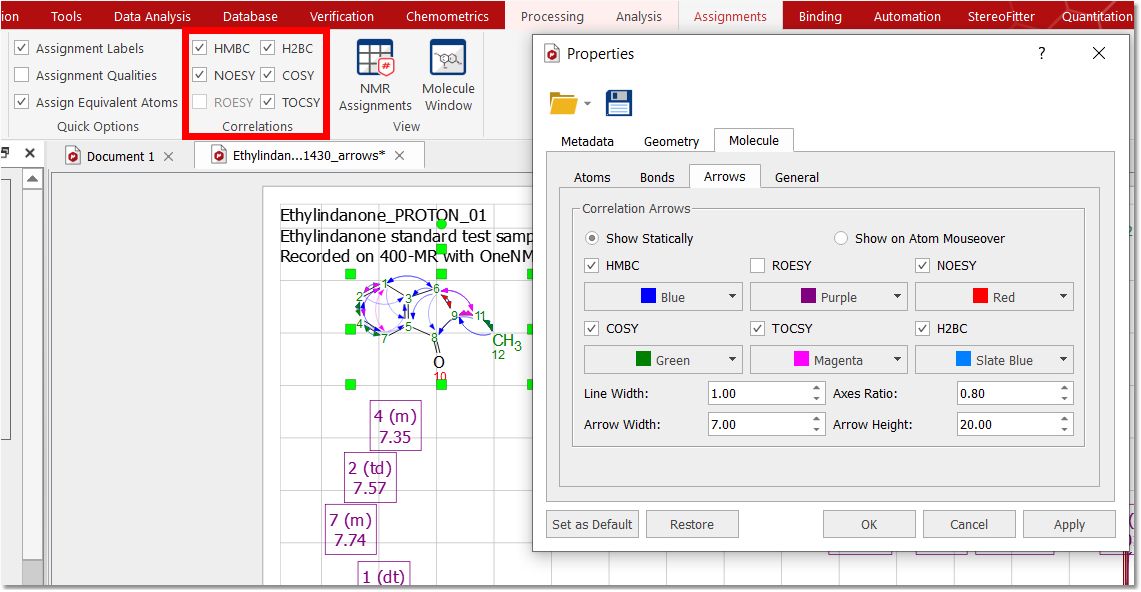
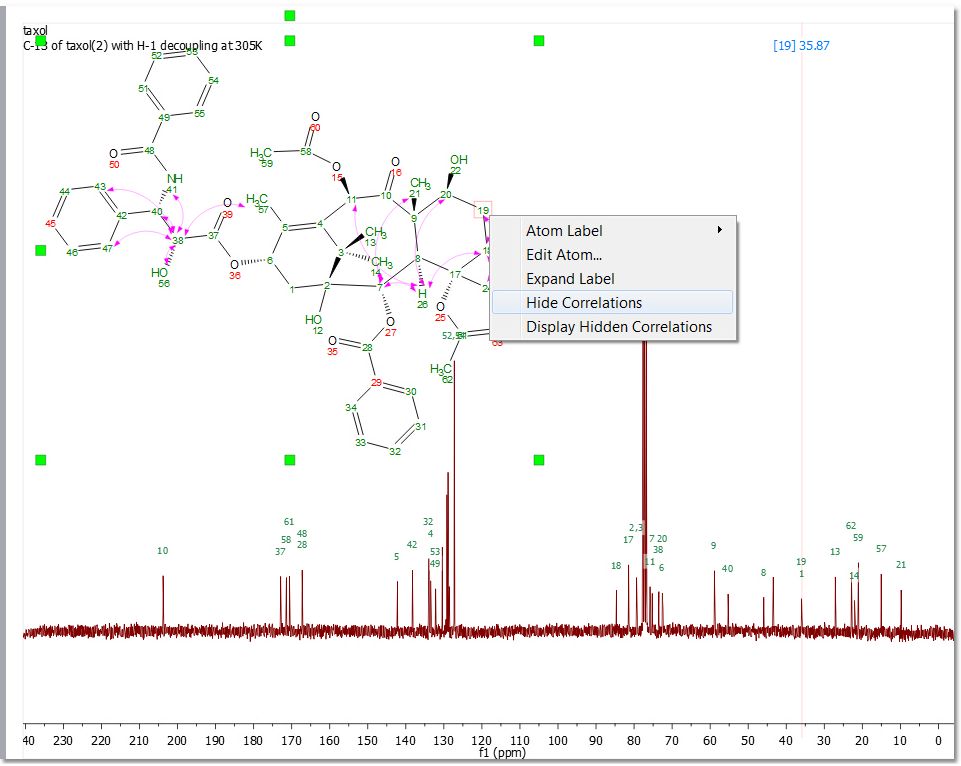
You can graphically visualize (by arrows between the coupling groups) the HMBC, ROESY, NOESY, COSY, TOCSY, H2BC and connectivity in the structure by checking the applicable boxes from the 'Assignments ribbon' or by double clicking on the molecular structure to display the 'Properties' and selecting the 'Arrows' tab to check the desired correlation arrows.
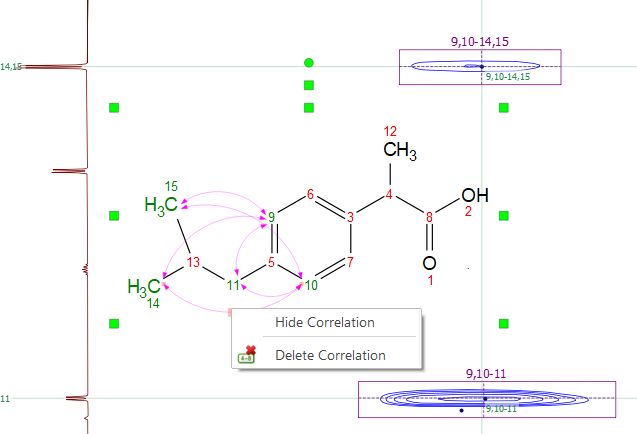
Hovering the mouse over the arrow and right clicking, will allow you to hide or delete the assignment:
The Assignments table shows checkboxes for each row in the correlations that can be displayed as arrows in the molecule. Checking/unchecking these checkboxes will change the visibility of the correlations for each atom.
Right clicking on the atom will allow you also to 'Hide Correlations'; you can show them again by using the command 'Display Hidden Correlations'.
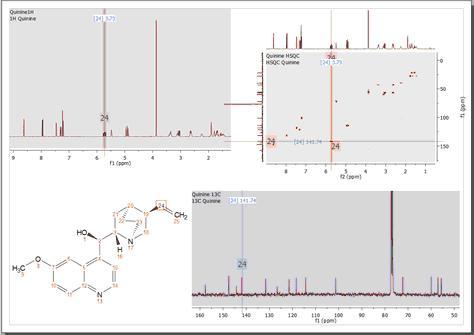
You can also copy several datasets in the same page (and after having resized them) to make the assignments. Here you can see a page which contains a 1H, 13C and HSQC datasets with an assignment in the molecular structure:
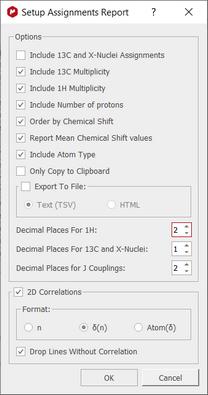
Click on the 'Report Assignments' button of follow the menu 'Tools/Report/Assignments' to export your assignments in journal format. Then, a new dialog will appear, to allow you to report the 2D correlations and the chemical shifts or to copy only the report to the clipboard (to paste it to another document) or export it as CSV or HTML file. From here you can also order by chemical shift, include 13C and X-nuclei assignments or multiplicity and report ranges or mean chemical shift values. When no chemical shift is included, the correlations get ordered according to the atom number. When chemical shifts are included, the correlations get ordered according to the chemical shifts values.
It is required at least a 1H spectrum. The table only will include columns for the spectra present in the document containing assignments. It generates a table in which each row corresponds to each non-equivalent proton.
Finally, you will get the report of the assignments pasted on the spectral window:
You can export the assignments as an ASCII file by following the menu 'File/Save As/Script:Assignments' or copy the assignments from one molecule to another (with the same numbering of the common parts).
You can also export your assignments as NMReData format by following the menu 'File/Save As/Script: NMReData (*.zip)'. The zip file will contain the raw dataset, the processing templates and a SDF file with the molecular structure and the assignments. Loading that zip file into Mnova will display your processed NMR datasets with the assigned molecular structure. |