Chemical Shift Referencing



Chemical Shift Referencing |
|
|
In NMR, the frequencies are usually measured relative to a frequency standard. As these frequencies are strictly measured relative to a frequency standard proportional to the magnetic field, it is convenient to define the chemical shift as:
Thus, in order to establish a chemical shift scale, it is necessary to choose some substance as a reference and define its chemical shift. In non-aqueous solvents, the most used reference substance is TMS (tetramethysilane) which is defined to have a chemical shift of 0.0 ppm. For biomolecules, slightly different compounds (e.g. TSP, (CH3)2SiCD3CD2CO2Na, Sodium-salt of trimethylsilyl-propionic acid) are used, since TMS is not soluble in water.
There are two way to reference your spectra, one graphically and another by using a dialog box.
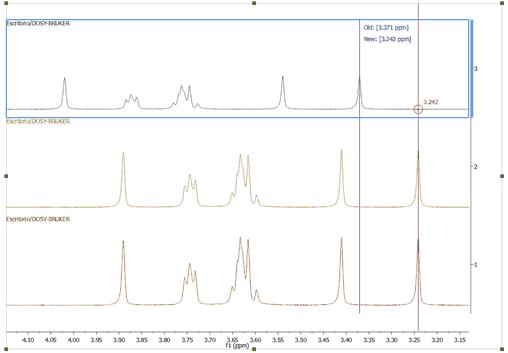
The Graphic Reference (R as shortcut) will allow you to reference your spectra just with two clicks. Once click to select the position that you want to reference and another click to select the new chemical shift. This feature will be very useful to reference stacked spectra (for example):
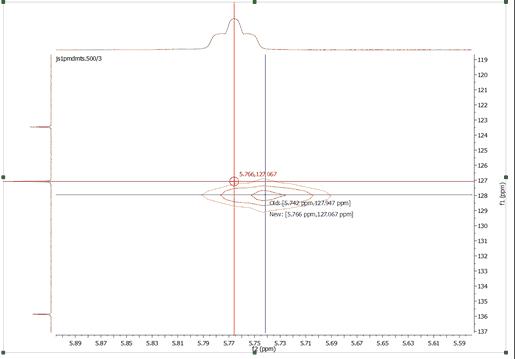
you can also use this tool to reference the 2D-NMR spectra directly with the 1D external trace. To do it just press the 'R' key, next click on the peak that you want to reference and finally click again on the new chemical shift (matching with the peaks of the external traces):
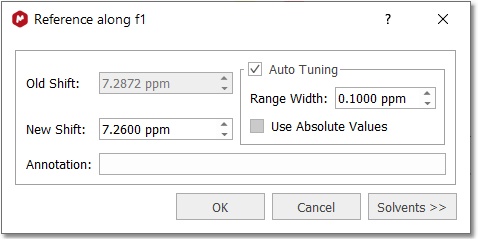
You can also calibrate your spectrum by selecting Analysis/Reference/References (L shortcut) on the main menu or by clicking on the
1. Zoom-in on the spectrum region containing the reference peak 2. Select the peak you want to be a reference point. You will notice that the mouse cursor automatically jumps to the peaks maxima. To select a peak at an arbitrary position which is not a local extreme (e.g. the shoulder of a peak), press SHIFT key, move the mouse to the desired position and click. The following dialog box appears:
You will be able to type any annotation after having referenced your signal.
The 'Auto Tuning' option finds the maximum peak inside a limit and references it. This is very useful for example if you need to reference automatically a large amount of metabonomic spectra, where the solvent peak could slightly change its shift with the pH of the sample. Check the '"use absolute values" to take into account also negative peaks for the reference.
You can type the new value for that peak or you can click on the
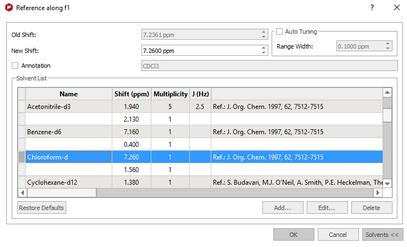
You can add new entries or edit the list of standard reference chemical shifts by using the buttons at the bottom of the dialog box.
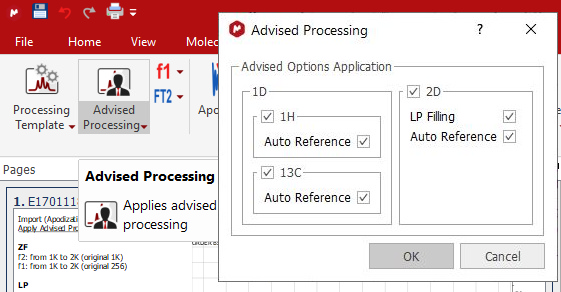
There is also an 'Advised processing' feature which will apply a suggested processing with the option to automatic reference the spectra:
|